


Summary
Prolonged or chronic critical illness – a term applied to patients that survive severe injury or infection, but fail to start recovering after a few days – is characterized by low levels of peripheral hormones (including T3, IGF-1, cortisol and testosterone). This pattern is increasingly recognized as a neuroendocrine dysfunction inhibiting patients’ recovery, requiring treatment independent of the initial illness or trauma.
The same pattern of altered hormone levels has been documented in ME/CFS and fibromyalgia, suggesting that the research into the mechanisms and treatments from the field of critical illness may be relevant for ME/CFS and fibromyalgia.
Pro-inflammatory cytokines play a role in inducing and maintaining the uniform suppression of the neuroendocrine axes — predominantly at the level of the hypothalamus — during prolonged critical illness irrespective of the initial injury or infection.
In order to remedy this neuroendocrine dysfunction in prolonged critical illness, treatments targeting different points along the various neuroendocrine axes have been trialed with some surprising successes. Some of the same treatments have also independently been trialed for ME/CFS and fibromyalgia.
Trials in prolonged critical illness, ME/CFS and fibromyalgia have shown benefits from direct supplementation with peripheral hormones (e.g. with T3, GH, IGF-1, cortisol and testosterone). Moreover, promising results from critical illness research suggests that treatments targeting the functioning of the central endocrine glands (i.e. the hypothalamus and pituitary) may be safer and even more effective for restoring normal metabolism than supplementation with peripheral hormones.
Table of Contents
- Critical illness: two different phases
- Neuroendocrine axes: regulated by central and peripheral mechanisms
- Hormones: a balance between catabolic and anabolic activities
Section 1: Neuroendocrine dysfunctions in critical illness and ME/CFS
- Normal conditions
- HPT dysfunctions in critical illness: non-thyroidal illness syndrome
- HPT dysfunctions in ME/CFS and fibromyalgia: low thyroid hormone activity
Somatotropic (growth hormone) axis: “HPS Axis”
- Normal conditions
- HPS dysfunctions in critical illness: loss of pulsatile function
- HPS dysfunctions in ME/CFS and fibromyalgia: low GH levels and failed response to exercise
Adreno-cortical axis: “HPA Axis”
- Normal conditions
- HPA dysfunction in critical illness: lower than expected cortisol levels
- HPA dysfunction in ME/CFS and fibromyalgia: depressed cortisol levels
- An Aside: the HPA axis “bi-stability” model
- Normal conditions
- HPG dysfunctions in critical illness: illness severity is correlated with axis suppression
- HPG dysfunctions in ME/CFS and fibromyalgia: higher scores of sexual dysfunctions
Interactions across the neuroendocrine axes and interactions with the immune system
Note: This is my second blog post on the relevance of research on critical illness for ME/CFS. I recommend you read my previous blog post first, but this post can also be read on its own.
Introduction
The neuroendocrine axes regulate every physiological process in the body, including metabolism, reproduction, growth, development, fluid balance, and the stress response.
Decades of research in the field of critical illness medicine have demonstrated that the neuroendocrine axes experience essentially the same profound alterations during all types of severe injury or infection – termed “critical illnesses.” However, the alterations differ significantly between the acute phase of critical illness (the first hours or days of onset) and the prolonged or chronic phase (when recovery does not occur within a few days) (van den Berghe, 2016).
Mechanisms involving pro-inflammatory cytokines and oxidative/nitrosative stress (O&NS) underpin these alterations (van den Berghe, 2000) and the subsequent mitochondrial damage that occurs (Preiser et al., 2014).
In parallel, decades of research have shown a suppression of neuroendocrine axes in ME/CFS and fibromyalgia (Riedel et al., 1998; Gupta and Silman, 2004), which, in fact, resembles that observed during prolonged critical illness. Moreover, patterns of pro-inflammatory cytokines and O&NS have also been documented in ME/CFS and fibromyalgia (Jason et al., 2009; Morris et al., 2016), and these have been shown to impact mitochondrial function and ATP production (Morris et al. 2019).
The objective of this blog post is to provide a summary of the neuroendocrine dysfunctions that typically occur during prolonged critical illness, and to highlight the similarities to dysfunctions documented in ME/CFS and fibromyalgia. I argue that the research from the field of critical illness may provide important insights into understanding the mechanisms of ME/CFS – and vice versa.
In this blog post I will:
- describe the alterations in the thyrotropic, somatotropic, adreno-cortical, and gonadotropic neuroendocrine axes during prolonged critical illness, as well as the similarities to findings in ME/CFS and fibromyalgia (Section 1).
- describe some of the interactions between the different axes (since the neuroendocrine system is best understood as a whole) (Annex)
But first, I will provide background on critical illness, the neuroendocrine axes and respective hormones.
Background
Note: This Background section is long, but important to understanding the neuroendocrine dysfunctions that occur during critical illness and in ME/CFS and fibromyalgia.
Critical illness: two different phases
Critical illness refers to the physiological response to severe injury or infection, such as sepsis, liver disease, HIV infection, head injury, pancreatitis, burns, cardiac surgery, etc. Independent of the underlying condition, critical illness is associated with an “excessive response” of pro-inflammatory cytokines (Marik, 2007) and is characterized by a uniform dysregulation of the neuroendocrine axes (Langouche et al., 2014). This dysregulation may be maintained even once the initial inflammatory surge has settled (Mesotten and van den Berghe, 2006).
Just like ME/CFS, critical illness is studied by looking at both the immune-inflammatory pathways (i.e. immune cells, cytokines, O&NS, etc.) and the alterations in neuroendocrine axes (i.e. the up- and down-regulation of hormone activity). The study of the interactions between the two (i.e. the immune-inflammatory pathways and neuroendocrine axes) is also increasingly a subject of critical illness research.
Severe infection or physical trauma can result in suppressed neuroendocrine axes that inhibit recovery; similar patterns are found in ME/CFS and fibromyalgia patients
[Source: https://websigns4u.com]
The two phases notably present “two distinct neuroendocrine paradigms” (Van den Berghe, 1998). Indeed, while the acute phase is characterized by increased release of pituitary hormones; the prolonged phase is characterized by a suppression of the release of pituitary hormones. Simultaneously, hormone circulation and hormone up-take by the peripheral tissues differ markedly between these two phases as well (Mesotten and van den Berghe, 2006). More details follow in Section 1.[1]
The acute phase is considered to be a beneficial or “adaptive” response to the severe stress of injury or infection (shifting energy and resources to essential organs and repair). However, the physiological mechanisms in the prolonged phase are now generally considered to be harmful or “maladaptive,” hindering recovery.[2]
Neuroendocrine axes: regulated by central and peripheral mechanisms
The neuroendocrine axes mentioned above are a series of glands that signal to each other to produce hormones. The main axes are the thyrotropic, somatotropic, adreno-cortical, gonadotropic and lactotropic axes, which respectively regulate thyroid hormone, growth hormone, adrenal hormones, gonadal hormones and prolactin (see Table 1).
For each of the axes, the hypothalamus (in the middle of the brain) sends signals to the pituitary (also in the brain). The pituitary, in turn, sends specific signals towards the various “peripheral” glands, including the thyroid (at the neck), the liver (behind the rib cage), the adrenals (above the kidneys), and the gonads (ovaries and testes). These peripheral glands then secrete a range of hormones that affect the genomic and metabolic activities of cells throughout the body[3].
Significantly, under normal conditions, negative feedback loops – acting just like a thermostat in a house – act on the hypothalamus and pituitary (i.e. the “central level”) to suppress further production of hormones and thereby maintain homeostasis. Thus, when plasma concentrations of the peripheral hormones reach a certain “set-level,” the neuroendocrine axes slow down production (see Figure 1).
Figure 1: The general cascade for production of peripheral hormones, and negative feedback loops
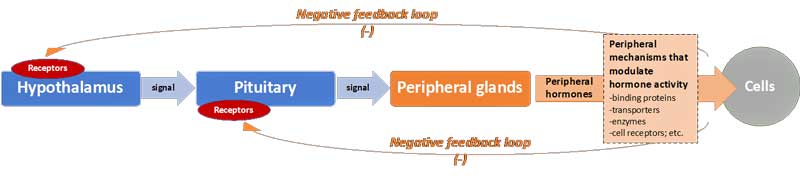
However, a number of “central” mechanisms can in fact alter the responsiveness of the hypothalamus and pituitary. These include the number and affinity (i.e. binding strength) of receptors for specific hormones in the two central glands. The more receptors, the more the hypothalamus and pituitary respond to signals. For example, if the number of hypothalamus receptors for cortisol increases, then it will slow down production at lower plasma cortisol concentrations (i.e. the hypothalamus has the impression that blood cortisol levels are elevated just because it has more cortisol receptors).
Additionally, mechanisms further “down-stream” (i.e. “peripheral” mechanisms) can also influence the level of hormone activity and (via the negative feedback loop) the overall function of the axes. They include:
- changes in the abundance and affinity of carrier molecules that bind specific hormones in the plasma making them more or less “available” to cells (c.f. “bioavalability” of hormones).
- increases/decreases in the energy-dependent transport of hormones into cells (and out of cells).
- relative up/down regulation of enzymes that convert hormones into active or inactive forms (specifically for thyroid hormones).
- variations in the form and quantity of hormone receptors at the level of the cells, thereby influencing the apparent “resistance” of cells to hormones.
- alterations in the rate of break-down of the hormones (and thus also their “half-life”).
- and others, not covered here.
Through experiments and observations, researchers are uncovering the complex combination of central and peripheral mechanisms that regulate neuroendocrine axes. Specifically, much research has gone into understanding how some physiological functions are put on hold (e.g. thinking and digestion) and others prioritized (e.g. combating infection or injury) during stress and critical illness (See Section 1). Crucially, researchers are finding that variations in these mechanisms are in part determined by the signals of the more than 100 different cytokines (van den Berghe, 2000).
One final relevant point regarding the function of endocrine axes: a lot of early research (and arguably clinical practice) in critical illness has been stumped by the failure to take into account the importance of the “pulsatility” of the signals from the pituitary to the peripheral glands (e.g. adrenals, gonads and liver). Indeed, the frequency and amplitude of the peaks and valleys in these signals (notably ACTH, LH and GH described in Section 1) may be as important as the overall volume of these signals in determining the production and activity of hormones (van den Berhge, 2000). However, pulsatility, of course, cannot be captured through just one blood test. Blood tests taken as frequently as every 20 minutes enabled researchers to capture signals’ peaks and valleys in order to demonstrate the endocrine dysfunction during prolonged critical illness (van den Berghe, 2016).
Hormones: a balance between catabolic and anabolic activities
The hormones produced by the neuroendocrine axes described above impact the activity of every cell. They determine our metabolism, growth, development, stress response, mood, strength, immune response, etc. Dysfunction in the respective axes producing these hormones leads to a myriad of symptoms present in prolonged critical illness, and possibly ME/CFS and fibromyalgia (see Table 1).
Researchers differentiate between anabolic hormones and catabolic hormones. Anabolic hormones (e.g. IGF-1 and testosterone) promote the building of molecules and tissue. The catabolic (e.g. cortisol and adrenaline) stimulate the break-down of molecules and tissues.
Researchers argue that catabolic activity may be beneficial during critical illness as amino acids, derived from the breakdown of peripheral tissues such as skeletal muscle and bone, are freed up for use by the central organs (Mesotten and van den Berghe, 2006). However, too many catabolic hormones relative to anabolic hormones in prolonged critical illness can lead to protein break-down in skeletal muscle, liver, kidney and heart, reducing their cell mass and leading to impaired function (Weekers and van den Berghe, 2004).
Similarly, the metabolic profiles of ME/CFS patients also exhibit a lack of anabolic activity (e.g. low NADPH) (Naviaux et al., 2016).
In sum: in just the last few decades, researchers have found that the neuroendocrine axes behave very differently in the acute vs the prolonged phases of critical illness, and that a multitude of central and peripheral mechanisms are involved. The result is a disbalance in the catabolic vs anabolic hormones, resulting in a myriad of symptoms during prolonged critical illness.
Table 1: Endocrine axes and function of the main hormones in adults
| Name of Axis | “Peripheral” endocrine glands | Main hormones | Function | Symptoms of suppressed hormone activity |
| Thyrotropic axis: “HPT Axis”
|
Thyroid gland | Thyroid hormones: T4, T3, T2, T1 and reverse T3 (RT3) | Regulate baseline level of metabolism
|
“Hypothyroid-like” symptoms: tiredness, stiffness, constipation, dry skin, etc. Weight gain. |
| Somatotropic axis: “HPS Axis” | Liver (mostly) | Growth hormone (GH) (produced by the pituitary) and IGF-1 (by the liver). | Regulation of insulin sensitivity, protein building (anabolic activity) and gut mucosal function | Low energy and weak muscle strength. Poor recovery after physical activity. Exhaustion. Anxiety. |
| Adreno-cortical axis: “HPA Axis” | Adrenal glands | Glucocorticoids, notably cortisol | Stress response via changes in glucose metabolism. Regulation of immune system | Inability to deal with stress. Proneness to exaggerated immune responses. Weight loss. |
| Mineralocorticoids, notably aldosterone | Regulate salt, water, electrolyte balance (blood pressure) | Low blood pressure. Dizzy on standing up. | ||
| Androgens, notably DHEA (can also be derived from HPG axis). | Function as steroids on muscle mass, fat storage, brain function, etc. | Muscle fatigue. Thinning & dry hair. Noise intolerance. | ||
| Gonadotropic axis: “HPG Axis” | Gonads (ovaries & testes) | Testosterone, estradiol, and progesterone | Sexual functions, moods, response to stress, muscle mass | Low testosterone: Weak muscle tone &
pain. |
| Lactotropic axis:
“HPP Axis” |
— | Prolactin (produced by the pituitary) | Role in metabolism and immune system (and milk production in women) |
Sources: Hertoghe, 2002; Gupta and Silman, 2004; Mesotten and van den Berghe, 2006; Elijah et al., 2011;
Section 1: Neuroendocrine dysfunctions in critical illness and ME/CFS
In this section I will describe the dysfunctions found in four of the major neuroendocrine axes during critical illness, and highlight the similarities to dysfunctions in ME/CFS and fibromyalgia. I will also summarize some of the “central” and “peripheral” mechanisms involved in these dysfunctions.
A. Thyrotropic axis: “HPT Axis”
The HPT axis regulates the basal rate of our metabolism according to a daily rhythm. I described the normal function and dysfunction the HPT axis during critical illness in a previous blog post, but I will summarize the information below.
Normal conditions
In normal conditions, a feedback loop works to maintain stable plasma thyroid hormone concentrations according to a daily rhythm (Fisher, 1996). In brief, when unbound thyroid hormone concentrations in the plasma dip below a certain threshold, the hypothalamus produces thyrotropin-releasing hormones (TRH) in order to signal the pituitary to produce thyroid stimulating hormone (TSH), which in turn signals the thyroid gland to produce more thyroid hormones (TH) (see Figure 2). Again, this resembles the function of a thermostat in a house.
Figure 2: The cascade for production of thyroid hormone (TH)

Once thyroid hormones are released by the thyroid gland into the plasma, further steps need to occur before they can impact the metabolism of the target tissue: they are carried around the plasma by thyroid binders, they are transported in and out of cells by cellular transporters, they are converted into the active or inactive forms by enzymes, and then are ultimately received by nuclear receptors in target cells initiating gene transcription[4].
HPT dysfunctions in critical illness: non-thyroidal illness syndrome
However, in the case of severe trauma or infection, this neuroendocrine axis doesn’t work as expected. This dysregulation has a name: non-thyroidal illness syndrome (NTIS) – also called “euthyroid sick syndrome” or “low T3 syndrome.”[5]
In brief, during critical illness the activities of enzymes responsible for the conversion of thyroid hormones into active and inactivated forms are altered: at the level of the liver (and other peripheral tissues) the enzymes are regulated to increase the conversion of thyroid hormones into their inactivated form (RT3); however at the level of the hypothalamus the enzymes are regulated to convert thyroid hormones into its active form (T3).
This results in a generalized higher ratio of inactivated thyroid hormone (RT3) to active thyroid hormone (T3) observable in blood tests; but a higher concentration of T3 at the level of the hypothalamus. As a result, the hypothalamus doesn’t signal the pituitary (and in turn the thyroid) to produce more thyroid hormones (i.e the axis is suppressed). Using the metaphor from above, it is as if the air around the thermostat was being heated, inducing the shutdown of the central heating.
These “central” and “peripheral” mechanisms translate into a general and tissue-specific down-regulation of metabolism. Researchers have found that these mechanisms are induced and mediated by cytokines and O&NS (see my previous blog post for details and references).
While NTIS was initially considered to be beneficial in critical illness – i.e. a state of “protective” down-regulation of metabolism during times of duress (Carter et al. 1974) – it is increasingly seen as “maladaptive” and hampering the recovery of patients in the case of prolonged critical illness (De Groot, 1999; Plikat et al., 2007; Boelen et al., 2011; Wajner et al., 2012; van den Berghe, 2016; Mancini et al., 2016; Chatzitomaris et al., 2017).
HPT dysfunctions in ME/CFS and fibromyalgia: low thyroid hormone activity
Since at least 1997 researchers have demonstrated that low thyroid hormone activity is also an underlying mechanism in ME/CFS and fibromyalgia (Lowe 1997; Lowe et al., 1998; Lowe, 2000; Lowe and Yellin, 2008). Last year, researchers found that CFS patients had a significantly higher ratio of RT3 to T3 hormones than controls (Ruiz-Núñez’s et al., 2018) which would also imply low thyroid hormone activity. They write: “low circulating T3 and the apparent shift from T3 to RT3 may reflect more severely depressed tissue T3 levels.” In other words, the neuroendocrine dysfunction of the HPT axis in ME/CFS patients resembles that of patients in critical care units[6].
In sum: the action of the thyroid hormone is depressed during prolonged critical illness. Similar patterns have been found in ME/CFS and fibromyalgia. Low thyroid hormone activity can result in a large variety of symptoms including fatigue. The concept of “non-thyroidal illness syndrome” recognized and studied in the field of critical medicine can likely inform the field of ME/CFS and fibromyalgia.
B. Somatotropic (growth hormone) axis: “HPS Axis”
The HPS axis plays important roles in growth and development of children, but also contributes to a variety of physiological pathways in adults, including balancing catabolic and anabolic activities (Mesotten and van den Berghe, 2006). HPS axis dysfunction is known to cause loss of muscle and bone mass, induce weakness (van den Berghe, 2016) and impact gut mucosa integrity as well as glucose and fat metabolism.
Normal conditions
Uniquely, in the case of the HPS axis, the hypothalamus sends both stimulating (+) and inhibiting (-) signals to the pituitary for the production of growth hormone (GH): these are, respectively, the GH-releasing hormone (GHRH) and the GH-inhibiting hormone (GHIH, also called somatostatin) (Mesotten and van den Berghe, 2006). In addition, ghrelin[7], mostly produced by the gut, also stimulates GH production by the pituitary (see Figure 3).
In normal conditions, GH is released by the pituitary in a pulsatile fashion under the control of these three signals, with peaks of GH levels alternating with virtually undetectable valleys in 3 to 5-hour intervals over the course of the day (van den Berghe, 2016). GH in turn has direct effects on some tissues and also stimulates the production of insulin-like growth hormone-1 (IGF-1) mostly by the liver. Nearly all of the IGF-1 hormones in the plasma are bound to IGF-binding proteins (IGFBP). IGF-1 and GH exert “negative feedback” on the hypothalamus and the pituitary to maintain homeostasis.
Figure 3: The cascade for the production of growth hormone (GH) and IGF-1
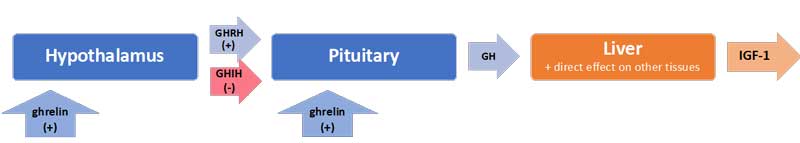
Interestingly, the half-life of GH is only 10 to 20 minutes, whereas the half-life of IGF-1 is more than 12 hours. Thus IGF-1 plasma concentrations are often used as proxies for GH secretion in clinical settings. However, GH and IGF-1 have different functions, particularly in the balance of anabolic vs. catabolic activities in the body. The use of IGF-1 as a proxy for GH ignores the impacts of the pulsatile secretion of GH on this balance.
HPS dysfunctions in critical illness: loss of pulsatile function
In the acute phase of critical illness, the pituitary produces more GH: higher peaks, lower valleys and increased pulse frequencies. This is, in fact, the result of the quick onset of two main peripheral mechanisms.
Firstly, under the influence of cytokines, the liver expresses fewer GH receptors (i.e. becomes “resistant” to GH) and thus produces less IGF-1 (van den Berghe, 2003). Secondly, alterations in IGF binding proteins results in IGF-1 being cleared out faster from the system (i.e. IGF-1 has a shorter “half-life”).
The lower IGF-1 concentrations resulting from these two peripheral mechanisms will – via the feedback loop inherent to the axis – spur more GH production (van den Berghe, 2016). Interestingly, recovery from critical illness is preceded by a normalization in IGF-1 and IGFBP levels (Mesotten and van den Berghe, 2006).
However, if a patient fails to recover within a few days, then GH secretion is no longer elevated (it may be low or normal), but the secretion becomes erratic and almost non-pulsatile (i.e. “loss of pulsatile function”). Experiments have demonstrated that this is largely due to a lack of stimulation by the hormone ghrelin – which you will recall is mostly produced by the gut (Mesotten and van den Berghe, 2006).[8]
As for the peripheral hormone, IGF-1, its levels are low or normal in prolonged critical illness. The “resistance” of the liver to GH (which had suppressed IGF-1 production during the acute phase of critical illness) does not persist during prolonged critical illness (van den Berghe, 2003; van den Berghe, 2016). However, without a concomitant pulsatile release of GH, the anabolic function of IGF-1 becomes inhibited (Mesotten and van den Berghe, 2006).
Again, although the increase in catabolic activity during the acute phase of critical illness may initially be beneficial because it serves to mobilize amino acids, the perpetuation of the imbalance in catabolic vs. anabolic activity (due in part to the loss of the pulsatile function of GH) is considered maladaptive. Thus, the dysfunction in the HPS axis contributes to muscle and bone wasting typically present in prolonged critical illness (Baxter 2001; van den Berghe et al., 1999).
HPS dysfunctions in ME/CFS and fibromyalgia: low GH levels and failed response to exercise
Dysfunctions in growth hormone regulation in ME/CFS and fibromyalgia have also been documented since at least the early 1990s. Indeed, a series of studies described GH to be relatively deficient in ME/CFS and fibromyalgia (Bennett et al., 1992; Bennett et al., 1997; and Allain et al., 1997; Berwaerts et al., 1998; Moorkens et al., 2000; Riedel et al., 2002; Paiva et al., 2002; Gupta and Silman, 2004; Cuatrecasas et al., 2010; Rigamonit et al., 2017).
Few if any studies appear to have assessed changes in the pulsatile function of growth hormone secretion in ME/CFS or fibromyalgia patients – probably due to the fact that this requires blood samples at frequent intervals day and night. However, studies have shown that fibromyalgia patients “failed to exhibit a GH response to exercise” (Paiva et al. 2002). In normal conditions, GH increases after exercise.
Similar to prolonged critical illness, IGF-1 levels are also documented to be generally low or low-normal in fibromyalgia patients (Bennet et al., 1992; Bennet et al., 1997; Bennet et al., 1998).
In sum: the pulsatile function of GH secretion is lost during prolonged critical illness. ME/CFS and fibromyalgia patients also exhibit alterations in GH secretion, including low GH and loss of response to exercise. Low or non-pulsatile GH secretion result in loss of muscle and bone mass, muscle weakness, and changes in glucose and fat metabolism.
C. Adreno-cortical axis: “HPA Axis”
The HPA axis is the body’s primary stress management system. The HPA axis responds to physical and mental challenge in part by controlling the body’s cortisol levels (Gupta et al., 2007). Cortisol in turn modulates inflammation response, cardiovascular function and metabolism (Téblick et al., 2019).
Normal conditions
In normal conditions, the adrenal gland secretes cortisol during the day in pulses, with the highest levels in the early morning hours and lower levels at night. The hypothalamus signals to the pituitary with corticotrophin-releasing hormone (CRH), and to a lesser extent arginine vasopressin (AVP), to produce adrenocorticotropic hormone (ACTH). This is in turn signals the adrenals to release cortisol (and other hormones). Most of cortisol circulating in the blood is bound to carrier molecules (Tomas et al., 2013; van den Berghe, 2016) (see Figure 4).
As in the other axes, the HPA axis also has a negative feedback loop. Specifically, when free circulating cortisol attaches to glucocorticoid receptors (GRs) on the hypothalamus and pituitary, these glands slow down production of CRH and AVP, and ACTH respectively. The number and affinity of GRs is thus considered one of the most important determining factors in the regulation of the HPA axis (Tomas et al., 2013).
Figure 4: The cascade for the production of cortisol

HPA dysfunction in critical illness: lower than expected cortisol levels
High cortisol concentration in acute illness and severe trauma is a vital response that allows for fluid retention, increased cardiac output and blood pressure, and induces an appropriate immune response while protecting against excessive inflammation (Boonen et al., 2015; van den Berghe, 2016).
This response was initially thought to be a result of the adrenals being fired up to produce more cortisol via the “central control” of the hypothalamus and the pituitary. Recently, however, it has become clear that the increased cortisol availability during acute critical illness is largely driven by two “peripheral” mechanisms: a decrease in the abundance and affinity of the cortisol binding proteins in circulation, and a slowing of cortisol breakdown in the liver and kidney. Together these peripheral mechanisms result in higher cortisol levels (Peeters et al., 2014; Boonen et al., 2015; van den Berghe, 2016; Peeters et al. 2018a; Teblick et al., 2019). In other words, the adrenals produce basically normal amounts of cortisol during acute stress, but peripheral mechanisms increase the action of the hormone by augmenting its “bioavailability” and lengthening its “half-life.”
The higher cortisol concentrations resulting from these “peripheral mechanisms,” however, suppress CRH, AVP and ACTH release via the negative feedback loop on the hypothalamus and pituitary. Without stimulation by ACTH, the adrenals produce less cortisol, and levels eventually drop.[9]
In cases of prolonged critical illness, ACTH levels (surprisingly) continue to be depressed despite a normalization in cortisol levels within 28 days of illness (Peeters et al., 2017; Peeters et al., 2018b). Why and how this central suppression of ACTH is maintained is not clear and continues to be debated. Pro-inflammatory cytokines and O&NS likely play a leading role (Marik et al., 2007; Boonen et al., 2015).
Indeed, there are several mechanisms by which inflammatory cytokines have been shown to impact all levels of the HPA axis. The cytokine IL-1β is known to modulate CRH release by the hypothalamus; TNF-a is known to impair ACTH release by the pituitary; and TNF-a is also known to impair cortisol production by the adrenal glands (Marik et al., 2007).
Crucially, cytokines can mediate tissue-specific changes in the abundance and affinity of glucocorticoid receptor (GR) – which, again, is a major factor determining the activity of the HPA axis (Marik et al., 2007; Boonen et al., 2015). It is as if the tables turn, and cytokines now have the “upper hand” over the regulation of cortisol (whereas usually we consider cortisol levels to be mediating the inflammatory pathways).
This shift may be partially explained by the fact that our adrenal glands atrophy if they don’t experience pulsatile ACTH stimulation for more than 1 week. (Recall that during the acute phase, ACTH release is suppressed due to a spike in cortisol resulting from the quick onset of peripheral mechanisms).
Indeed, clinicians have observed adrenal atrophy and loss of integrity (e.g. zonational structure) in the post-mortem dissection of patients that had been critically ill for a few weeks, but not in the patients that quickly died from their illness or trauma (Boonen et al., 2015; Peeters et al. 2018a; Peeters et al., 2018b; Teblick et al., 2019). If the adrenals are thus weakened, it may be difficult for the HPA axis to regain the “upper hand” over the immune system during prolonged critical illness.
HPA dysfunction in ME/CFS and fibromyalgia: depressed cortisol levels
Dysfunction of the HPA axis has also been documented extensively in ME/CFS patients at least since the early 1990s (Demitrack et al., 1991; Scott et al., 1998; De Beeker et al., 1999; Gaab et al., 2002; Crofford et al., 2004; Jerjes et al., 2005; Segal et al., 2005; Van Den Eede et al., 2007; Van Den Eede et al., 2008).
Researchers have observed decreased baseline cortisol levels, blunted HPA axis responses to physical and psychological stressors, reduced HPA-axis responsivity to provocation tests (such as CRH and ACTH administration), and a heightened negative feedback loop (consistent with a higher abundance and affinity of glucocorticoid receptors at the level of the pituitary and hypothalamus).
Moreover, the morning peak of ACTH which stimulates the adrenal glands, is often missing or weak in ME/CFS patients, and the lack of pulsatile stimulus by ACTH also leads to adrenal atrophy in these patients (Scott et al., 1999). In other words, the HPA dysfunction in ME/CFS is not unlike the dysfunction in prolonged critical illness.
Strikingly, the magnitude of HPA axis dysfunction becomes more pronounced with illness duration and is associated with symptom severity (see review in Tomas et al., 2013). Additionally, peripheral mechanisms that depress the HPA axis such as increased levels of cortisol binding globulin, CBG (which decreases cortisol availability to the tissues) have been observed in CFS patients.
Similarly, HPA axis dysfunction is also present in the majority of fibromyalgia patients (Kirnap et al., 2001; Riedel et al., 2002; see review in Holtorf, 2008). Various mechanisms have been suggested, including depressed secretion of CRH by the hypothalamus, a deficiency of CRH receptors on the pituitary, and adrenal atrophy due to chronic under-stimulation by reduced ACTH levels (Gupta and Silman, 2004).
Undoubtedly the HPA-axis is suppressed in ME/CFS and fibromyalgia, and this is associated with pro-inflammatory cytokines and O&NS (De Beeker et al. 1999; Jason et al., 2009; Tomas et al., 2013; Morris et al., 2014; Morris et al., 2016). However the “bidirectional relationship” between the HPA-axis and inflammation (i.e. cortisol regulates the immune system; and inflammation can impact the HPA-axis) has lead researchers to struggle with the question of causality.
Specifically, researchers have pondered whether the down-regulation of the HPA-axis “permits” the excessive inflammatory pathways or whether causality runs the other way: i.e. if the inflammatory pathways are suppressing the HPA-axis (Tomas et al., 2013).
The conclusion of a recent paper that immune-inflammatory and O&NS pathways induce HPA axis dysfunction in ME/CFS (Morris et al., 2016) is also consistent with the observation that the inflammatory pathways induce endocrine dysfunctions in critical illness (independent of the nature of the original severe injury or infection).
In sum: the HPA-axis is suppressed in prolonged critical illness, as well as in ME/CFS and fibromyalgia. Low cortisol levels result in an inability to deal with stressors and a proneness to exaggerated inflammatory responses.
An Aside: the HPA axis “bi-stability” model
ME/CFS researchers have developed a fascinating model to explain the persistence of a suppressed HPA-axis (Gupta et al., 2007; Ben-Zvi et al., 2009). The model suggests two “stable steady states” of cortisol production: a high and low state. Researchers call this the “bi-stability of the HPA axis.” In other words, the body (through a network of feed-back mechanisms and other physiological mechanisms) will always tend towards one of these two steady states.
The model is based on the fact that stress (via cortisol) influences the number of glucocorticoid receptors (GRs) on the hypothalamus and pituitary. Recall that GRs play an essential part in the negative feedback loop: when cortisol from the plasma binds to them, they inhibit the production of CRH by the hypothalamus and ACTH by the pituitary (like a thermostat sensing that the house is warm enough). Thus, the higher the number of GRs, the less CRH and ACTH secretion; this ultimately leads to reduced cortisol production by the adrenals further down the axis[10].
The researchers argue that a short stress (i.e. a burst of cortisol) will produce a small perturbation in the concentration of GRs on the central glands which quickly returns to normal levels. However, long, repeated stress – from which the system doesn’t have time to recover – leads to a persistent state of high levels of GRs, forcing the HPA axis to an alternate ‘‘low cortisol” steady state. In model-speak: “the HPA axis reached the basin of attraction of the second stable steady state and remained there even after the removal of stress” (Gupta et al. 2007).
More recent models of the HPA axis have also included “non-genomic” feedback-controls (Zarzer et al. 2013), the endogenous effects of circadian rhythm (Hosseinichimeh et al., 2015), and interactions with the HPG axis and the immune system (Craddock et al., 2015; Morris et al., 2019) to explain how patients can get stuck in a “low-cortisol” steady state (i.e. how HPA-axis suppression is maintained even after the initial stress is gone).
D. Gonadotropic axis: “HPG Axis”
The HPG axis not only regulates the reproductive system, but also impacts our immune system, muscle development and metabolism.
Normal conditions
Similar to other neuroendocrine axes, the production of gonadal hormones (i.e. estrogen, progesterone and testosterone) is regulated by the hypothalamus via the gonadal releasing hormones (GnRH) which stimulates the release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) by the pituitary.
These signals in turn stimulate the gonads (i.e. ovaries in women and testes in men) to produce estrogen, progesterone and testosterone. The system is kept in check via negative feedback loops on the pituitary and hypothalamus (see Figure 5).
Figure 5: The cascade for the production of gonadal hormones

HPG dysfunctions in critical illness: illness severity is correlated with axis suppression
The HPG axis response to critical illness has been studied much less than the other axes. However, existing studies show suppression of the HPG axis occur in both acute and prolonged critical illness states. Moreover, the more severe the illness, the greater the FSH suppression appears to be, implying that fewer signals are transmitted to the gonads to produce hormones (Raj et al., 2016; Vaikkakara et al., 2017). Indeed, testosterone levels are extremely low during prolonged critical illness in men (van den Berghe et al. 2001).
Similar to the other axes, researchers also surmise that pro-inflammatory cytokines are the probable mediators of the HPG axis suppression, via their inhibitory impact on the pulse of GnRH release by the hypothalamus (van den Berghe et al. 2001; Raj et al., 2016)[11].
HPG dysfunctions in ME/CFS and fibromyalgia: higher scores of sexual dysfunctions
There are also far fewer studies on the HPG axis in ME/CFS and fibromyalgia patients than on the other axes.
Higher scores of sexual dysfunctions were observed in fibromyalgia patients (Alves et al., 2016); and earlier menopause in ME/CFS patients (Boneva et al., 2015). Lower estrogen levels were also observed in fibromyalgia patients than in controls (Gupta and Silman, 2004). Moreover, it was determined that the cells of the immune system of ME/CFS patients have significantly lower estrogen receptors (Gräns et al. 2006), possibly inhibiting the effect of estrogen in modulating the immune system in ME/CFS.
Studies have also shown that levels of progesterone and testosterone (in fact derivatives of adrenal hormones in women) relate to pain sensitivity in fibromyalgia patients (Schertzinger, et al., 2018).
In sum: the HPG axis is suppressed during prolonged critical illness. Abnormalities related to gonadal hormones are also observed in ME/CFS and fibromyalgia. Muscle weakness and pain sensitivity can be a function of altered progesterone and testosterone levels.
E. Interactions across the neuroendocrine axes and interactions with the immune system
The neuroendocrine axes are often studied and described in isolation in both the literature on critical illness, and the literature on ME/CFS and fibromyalgia. This is problematic, however, because the various axes interact with each other – as well as with the immune system. (see Annex).
Whereas each of the neuroendocrine axis would tend to revert the body to a healthy homeostasis via feedback loops, the sum of the interactions across neuroendocrine axes — and interactions of neuroendocrine axes with the immune system — could create alternative and persistent “hypo-metabolic” steady states.
Certainly, if we had a comprehensive model of the various neuroendocrine axes – including how they interact with each other as well as with the inflammatory pathways – we might be able to better understand both critical illness and ME/CFS.[12] And we might also know what levers to use to move the whole system back to a normal state.
However, even based on current levels of knowledge, researchers on critical illness and ME/CFS have had some surprising successes with trial treatments that take into account the mechanisms within and interactions across neuroendocrine axes, as well as with the immune system (see Section 2).
Section 1 Summary:
Prolonged critical illness, ME/CFS and fibromyalgia are characterized by neuroendocrine dysfunctions (see Table 2). Research on critical illness has revealed that the suppression of the neuroendocrine axes mostly originates at the level of the hypothalamus during prolonged critical illness, and that inflammatory pathways play a role in inducing and maintaining this suppression.
These mechanisms may also explain the neuroendocrine dysfunctions found in ME/CFS and fibromyalgia. Such neuroendocrine dysfunctions have wide ranging implications, including on the balance between anabolic and catabolic processes, metabolism, and the regulation of the immune system.
Table 2: Summary of neuroendocrine dysfunctions and mechanisms in critical illness and ME/CFS
| Dysfunctions
Critical illness & ME/CFS (+ FMS) |
Mechanisms
in prolonged critical illness |
|
| HPT Axis | Low thyroid hormone function
-Lower T3 -(Lower T4) -Higher RT3:T3
|
Hypothalamus: cytokine-induced alteration in set-point for release of TRH
Pituitary: cytokine-mediated suppression of TSH secretion Thyroid gland: cytokine-mediated reduction in T4 secretion by the thyroid gland. Periphery: Upregulation of T3 to RT3 conversion (notably in liver), etc. |
| HPS Axis
|
Low growth hormone function
-Loss of pulsatile release (in prolonged critical illness) -Lower GH secretion (in ME/CFS and fibromyalgia) -Low or normal IGF-1 |
Hypothalamus: lack of stimulation by gherlin; change in relative balance of hypothalamic stimulating / inhibiting hormones (GHRH / GHIH)
Pituitary: lack of stimulation by gherlin Liver: reduction in GH receptors (resistance to GH), but apparently only during acute phase |
| HPA Axis | Lower than expected cortisol function
-Lower cortisol baseline than expected -Blunted HPA axis response to stressors -Increased negative feedback -Loss of morning ACTH peak |
Hypothalamus: cytokine-mediated increase in glucorticoid receptors (which inhibit CRH release)
Pituitary: increase in glucorticoid receptors; decrease in CRH receptors (inhibit ACTH release) Adrenal gland: adrenal atrophy (due to lack of pulsatile ACTH stimulation during acute phase) |
| HPG Axis | Suppression of HPG axis (in critical illness)
(unclear for ME/CFS and fibromyalgia) -Lower levels of testosterone -Sexual dysfunctions |
Hypothalamus: cytokine-induced suppression of pulse of GnRH release |
The Low T3 Series on Health Rising
- The Atypical Thyroid Issues in Chronic Fatigue Syndrome (ME/CFS), Plus a New Thyroid Subset?
- Pure T3 Thyroid and Stories of Recovery from Chronic Fatigue Syndrome (ME/CFS) and Fibromyalgia: An Overview.
The Critical Illness Series On Health Rising
- Neither dying, nor recovering”: Learning from ICUs to Solve ME/CFS and Fibromyalgia – A Synopsis (Nov. 2019)
Notes
[1] Note: This “biphasic” pattern of the neuroendocrine system during critical illness was really only recognized in the early 1990s (Weekers and van den Berghe, 2004)!
[2] Note: Researchers surmise that because prolonged critical illness basically did not exist prior to the development of modern intensive care medicine (i.e. individuals would not have survived), the processes in this phase were not subject to evolutionary forces (Weekers and van den Berghe, 2004).
[3] Note: signals sent by the hypothalamus and pituitary are tropic hormones; peripheral glands produce non-tropic hormones (i.e. peripheral hormones).
[4] Note: thyroid hormones can also exert non-genomic effects
[5] Note: NTIS is not “hypothyroidism.” Indeed, the terms “non-thyroidal” and “euthyroid” indicate that that the dysfunction is not at the level of the thyroid gland and that the signal from the pituitary (i.e. TSH) is within the normal range.
[6] Note: A further neuroendocrine dysfunction implicating the immune system has also been repeatedly documented in fibromyalgia patients: higher rates of antithyroid antibodies – also called Hashimoto’s (Lowe and Yellin, 2008).
[7] Note: gherlin was only discovered in 1996 and first reported in 1999.
[8] There is also evidence for changes in the relative amounts of GHIH and GHRH signals from the hypothalamus (cite).
[9] Note: circulating bile acids also appear to play a role in CRH suppression
[10] Note: cortisol also inhibits the expression of CRH receptors on the pituitary; with less CRH receptors the signal from the hypothalamus to the pituitary is blunted.
[11] Note: estradiol concentrations are increased in both men and women during critical illness, but those are likely products of the conversion of androgen hormones secreted by the adrenals, not products of the gonads (van den Berhge, 2000).
[12] Note: the most comprehensive model I found was by Morris et al., 2019
Annex: Some interactions between neuroendocrine axes and interactions of neuroendocrine axes with the immune system.

This work is licensed under a Creative Commons Attribution 4.0 International License.



Hi Cort,
Thanks for having the courage for tackling such a complex topic. I had to read it diagonally and zoom in on things to get trough it, let alone writing it. I will have to reread it later, maybe a few times.
So far I wonder if we don’t look at both Prolonged Critical Illness, ME and FM with modern day eyes. Most of humans history, people lived nomadic. There was seldom enough food for a group to stay at a single place for more then a few days.
When you got ill, you had to recover sufficiently in a few days and be able to keep up with the group or to be left behind. When left behind, chances to recover and be able to have children to pass on your genes were slim. So having a few days “giving your all” to recover was kind of a smart genetic adaptation.
When left behind, you probably had better chances “sitting it out”, reduce activity and break down less essential parts of your body in order to try and heal. It probably was better to hide from dangers and wait and hope then to try and catch up with the group.
So maybe the few that passed on their genes after not recovering in days from acute illness were indeed those who could wind themselves down, having the feeling and believe they had too few energy to try and catch up with the group and who lacked the mental drive to try and push through against all odds.
Near all sick animals show the same behavior when being sick: lie down, rest and hide till they either die or get better. Probably this is because it offers the best chances to make it through.
Inhibiting metabolism and living on reserves by breaking the body down (catabolism) rather then expose oneself and trying to gather food while limping may have been the natural way to survive and have been a more successful adaption rather then a mal-adaptation. Who recovered this way within two or three weeks could survive for some time after on his own and join the next group passing by.
The one trying to be as active as possible and try to catch up with the group may have ever failed to do so and to recover in order to stay with the group. Maybe a better historical understanding what our bodies are trying to do could lead to better treatments?
@Cort: Most people think “negative feedback” is bad and positive feedback is good, while negative feedback is stable (most often better) feedback and positive feedback is unstable (most often bad) feedback. As you use the term several times it may help to clarify this.
These musings are fascinating. I can’t help but think an indefinite, inescapable viscious cycle leading to near incapacitation seems as likely to have reduced the mating prospects of my ancestor on the Serengeti as it is reducing mine in 2019. But evolution is a work in progress, and any given adaptation/maladaptation we analyze is really the aggregate of thousands of tiny random variations that, together, budge probability of gene propagation in one direction or the other. It could be that many aspects of the underlying cause of ME/CFS are actually adaptive (which is why it’s reached present day) but we’re missing one key piece that might have allowed us to escape from the cycle after several days of rest rather than several decades.
@Dominic
Thanks for all the detailed work. I saw too late it was your writing. My apologies.
Does anyone know any ME/CFS clinicians who have any focus on this aspect of treatment? I was found to have hypothyroidism years after first being diagnosed with ME/CFS, and have had issues with adrenals as well. I find that the doctors who treat those specific endocrine issues think it will solve everything, though, and get confused when my body doesn’t react quite like they’re expecting to treatment. Would really appreciate locating a doctor who focuses on ME/CFS and also understands the endocrine pieces of treatment. Thanks for any leads!
I dutifully take the very expensive Armour thyroid because my endocrinologist says it will help my thyroid (Hashimoto’s). I don’t think it makes me feel any better, but I’m assuming it’s protecting me from the thyroid gland getting “sick” (e.g., nodules, goiter, etc). I trust him because he goes about things in a very scientific and compassionate manner. But he lets my CFS doctor deal with the CFS aspects. Just as we need to find multiple therapies, we probably need to find multiple doctors. But good luck.
PS to Lisa – I think it’s very common for people with ME/CFS to have Hashimoto’s Disease (autoimmune to thyroid) and other thyroid disorders. Would be great if just taking thyroid hormone would restore health, but not so easy. I gather that most CFS doctors realize the thyroid connection; endocrinologists are probably are more conservative bunch. At least, that’s been my experience. My CFS doctor is an immunologist (but they can also be a conservative lot). I hope you’re near enough to one of the urban centers with a name CFS doc that you can at least have a consultation and long distance treatment from them. I wouldn’t try to “educate” the endocrinologist. They’re good at what they do, and that’s what they do. Best of luck.
Could all this be confusing because we are being poisoned ….our foods , medicine ,hygiene, ,addiction…..etc all pose serious health issues ……Most everything listed in this endocrine system illness might be Mineral Deficiency issues ….Question …? Does our use of GMO foods loaded with Glyphosate ….could this chemical cause Mineral Disruption of needed minerals ….We are all mineral deficient ….we need about a dozen minerals and calcium is not very important ….There are two most important minerals that you have been brainwashed into believing they are toxic …..Keep searching …..The reason you are ill is from the terrible three …bromide , fluoride , and chloride …..The thyroid sucks these up causing illness ……keep searching ……
John, your “terrible three” and the “-ine” versions of them, are everywhere. swimming pools, drinking water, hygiene products, etc. When I was a kid in the 70’s, our elementary school was part of an experiment, as an empoverished school district, where the kids had to eat fluoride tablets. I remember hearing the janitors had had enough of cleaning the pills out of the drinking fountains, and the school stopped the project. Anyway, that was only the beginning of the assault on the innocent in this country.
I have not understood for a long time what the difference is between ME/CFS, adrenal fatigue, adrenal insufficiency/failure eventually, GWI, fibromyalgia, burnout, nervous breakdown. They all seem pretty much very similar. The medical community has refused to acknowledge there can be a problem with people’s adrenals, do not even have a category for it unless you have developed Addison’s disease, may actually get reprimanded if they use the term Adrenal fatigue; in all the terms above, the medical community has continued to say it’s in people heads (until recently when they can’t look the other way any longer with so many people developing these conditions). Can’t the adrenal system breakdown like any other system in the body when confronted with unrelenting stress (physical, emotional, environmental, psychological, etc). You reach your breaking point (everyone reaches it differently based on your own bodies strengths/weaknesses/reserves etc), and then it manifests a little different in everyone because of this also. (Loss of hormones at mid-life, brewing hidden infections, personality type, heavy metal exposures, enormous stress for all just going thru daily grind nowadays, glycosphate poisoning our food supply/body burden, EMF, drinking recycled waste for water (don’t understand why they can’t get salt out of sea water for clean drinking water but that’s another issue to tackle at some point), I don’t know but seems like the condition the medical community refuses to acknowledge (adrenal fatigue/insufficiency) and therefore haven’t put any effort forth in studying could very well be the thing we’re all struggling to figure out and get well from. Can someone please tell me what the major difference is in all of these in simple terms.
Ghrelin is the correct spelling, not gherlin.
Fixed – thanks.
Having years of trying to figure out my WHYS, I have many of my puzzle pieces. Back in my early 30s a Functional medicine doc figured out I had issues with T4 to T3 and RT3 issues. We tried reversing this and upping my super low body temperature with T3 titrating therapy. This was not a good thing to try. We never got my body temperature up and it was a very hard on you. Gave worse issues with blood pressure and tachycardia. (Didnt know I had HyperPOTS then.)
He discovered I had low cortisol. We tried giving supplemental cortisol. That didn’t help and I got even more dysfunctional and tired. But, I still have to use cortisol with surgeries last I have a complete adrenal crash.
Then he checked Growth Hormone and found that very low. My insurance wouldn’t cover a trial of that. So we addressed it with homeopathic medicine. Couldn’t tell much diffence with that either.
Then was found super low aldosterone and renin. With these two low, treatment is usually high diuretics. Nephrologist said that wasn’t even to be considered with my having POTS and low blood volume. Treatment to try to up aldosterone and improve POTS, was horrible for me. Upping salt so wrong with my issues.
Now my doc is saying my pituitary isn’t producing correctly. We are trying a nasal hormone for that. Seems to be helping energy.
Then there is balance between sex hormones. I do think there is an estrogen dominance issue. Both for men and women. Having had endometrosis and a full complete hysterectomy at a very young I wonder if there wouldn’t had been cancer if I hadn’t had to do that so young. My dad had prostate cancer and that predisposes daughters to gynecologic cancers. I’ve asked several men who had both POTS and CFS if they had the balance between testerone and estrogen checked. Some had and there was an issue. So may be one key and a reason why most CFS and POTS is female dominant and why some men may have issues.
I have no doubt that the endocrine system is out of balance. But my experience of trying to correct these things all fell short and I did not get corrections.
It may be some upregulating and downregulating as an attempt to correct something else of more importance. It could be compensations. Man knows alot about the function of the body, but new light comes forth daily.
I’m still alive and that first discovery of endocrine dysfunction was 30 years ago. Still chronic, but still searching for the elusive “purple bandaid”.
We are all so complex.
Issie, thanks that out in detail. I, too, have been looking for that purple band-aid for so long that I don’t know what I would do with myself if I ever get there. My doc suspects brain endocrine problem, too.
I didn’t even read the article yet because it’s so complex. But this stuff is so important. Thanks for posting Cort.
Boy are you a good example of how complex these issues are!
I was kind of stunned reading Dominic’s article how complex they are (and that he was able to wrap them all together.) I now feel for endocrinologists trying to stabilize or fix these intertwining systems.
Yes, especially when there are so many dysfunction and other known DX won’t allow treatment as it would make them worse.
I’ve done alot of experiments and doctors have learned as we try and fail. But we get more knowledge.
Issie
Well done, Dominic. A lot of hard work for you but a much needed focus on the endocrine system which I have always thought to be the most important aspect of these illnesses. There has been too much focus on the immune system which, of course, will be affected by a dysfunctional endocrine system.
The endocrine system was a big focus at one time and then faded but particularly with Dr. Klimas’s work, Cortene’s drug and the Metabolic hypothesis – it’s come back. Her computational biology modeling efforts – which Dominic alludes to in the next blog in the series – indicate that issues with the HPA axis MUST be addressed.
https://www.healthrising.org/blog/2019/09/06/neuroendocrine-critical-illness-chronic-fatigue-fibromyalgia-treatment/
Yes, you are right, Cort. The HPA axis dysfunction must be addressed but we have all known this for more than 20 years. It was an early finding but how much closer are we to fixing it? Up until now it has been ignored that all the endocrine axes are inter-dependent. It maybe that the only axis that gives no symptoms turns out to be the most important as despite the fact that little is known about prolactin, it is generally believed that it is a moderator of the HPA axis and a promoter of neurogenesis. The fact that these illnesses are in the female majority connects, as prolactin is more important in women as it produces breast milk as well as preparing the body for pregnancy. It’s production is regulated by dopamine, something we are short of in these illnesses, causing high prolactin levels. Probably why cannabis helps so much. The other interesting fact is that there is a belief that the stress response is different between males and females. When females are stressed their cortisol goes down so it could be that it is prolactin that is causing this down-regulation of the HPA axis.
You give any endocrinologist a run for their money ????
I love the level of complexity even if my fogged brain have some challange understandig everything. This gives the page credbility and makes it possible to recommend your page to medical practitioners.
Onset for me wasnt flu, but hyperparathyriodism. High levels of calsuim over time, played probably a role in nevrological sympthoms.
I find these connections hard to understand, but very pausible. I know f.ex no woman with ME that doesn’t get tremendously worse when PMS or postmenopause-periode.
I wish for many studies focusing on neuroendocrinologys role in ME.
Thanks again for Your GREAT work!
F
“You give any endocrinologist a run for their money ????”
I suspect that may be true and that doctors who know the endocrine system would embrace Dominic’s work. Congrats to him for making his way through such a complex subject.
Thanks for posting this Dominic. I am so glad, after being treated like a liar all my life, that research and science and docs out there who taking this serious after all these years.
This article hits home for me. Lupron (a gnrh drug) was the cause of my chronic illnesses. It altered the HPG axis and I never produced estrogen or progesterone after Lupron. I fully believe, that at least for me, HPG axis dysfunction is the cause of my illnesses.