(We’re in the windup phase of Marco’s Neuroinflammatory Series as he explores the role the immune system and pathogens play in producing the neuroinflammatory state that may underlie chronic fatigue syndrome (ME/CFS)
C – IMMUNE FINDINGS, ETIOLOGY AND ONSET
Last time out I set out to show how a neuroinflammatory model could explain the major, minor and overlap (or even unreported) symptoms of ME/CFS. This time I’ll try to reconcile the model with the immune findings and type of onset.
Immune findings
Numerous immune abnormalities have been found in ME/CFS cohorts but findings are often inconsistent or even at times contradictory, are non-specific to ME/CFS and therefore not useful as a ‘biomarker’.
Nevertheless immune dysregulation of some sort does appear to be a consistent feature of ME/CFS although it has yet to be determined if the immune system dysregulation is a key driver or a downstream effect. Some immune findings suggest past or current viral infection involving a range of pathogens, i.e. no one pathogen has been implicated.
Consistent Findings
Amongst the few consistent findings are impaired NK cell cytotoxicity (Brenu et al, 2011) and increased pro-inflammatory cytokines such as TNF-alpha (Brenu et al 2011, Maes et al, 2012)
natural killer cell receptor
Except for the natural killer cell findings the immune findings haven’t been dramatic (but see Broderick’s work suggesting that studying immune networks is more effective than studying immune factors one by one.)
In a review of immune findings in ME/CFS to date, Bansal et all, 2011 concluded:
“Despite many years of intense investigation there is little consensus on the presence, nature and degree of immune dysfunction in this condition. However, slightly increased parameters of inflammation and pro-inflammatory cytokines such as interleukin (IL) 1, IL6 and tumour necrosis factor (TNF) α are likely present. Additionally, impaired natural killer cell function appears evident.”
Immune markers of inflammation
These findings are remarkably similar to Danzer et al’s description (discussed in Part I) of common immune markers found in “medically ill patients with a range of conditions in which inflammation played a key role”. Dantzer noted that “especially hs-CRP and IL-6 have been found to reproducibly identify the presence of an activated innate immune response”
High-sensitivity c-reactive protein (hs-CRP) is a common and non-specific marker for systemic inflammation which appears to be raised in ME/CFS (Raison et al, 2009).
“ Log-transformed mean plasma concentrations of hs-CRP were increased in subjects with (ME)CFS when compared to subjects who were well”
You may also recall that Rönnbäck and Hansson’s description of ‘mental fatigue’ discussed in Part II suggested the role that pro-inflammatory cytokines may play in mental fatigue :
“we present the hypothesis that the proinflammatory cytokines tumor necrosis factor-α, IL-1β and IL-6 could be involved in the pathophysiology of mental fatigue through their ability to attenuate the astroglial clearance of extracellular glutamate, their disintegration of the blood brain barrier, and effects on astroglial metabolism and metabolic supply for the neurons, thereby attenuating glutamate transmission.”
Primary biliary cirrhosis as a model for ME/CFS?
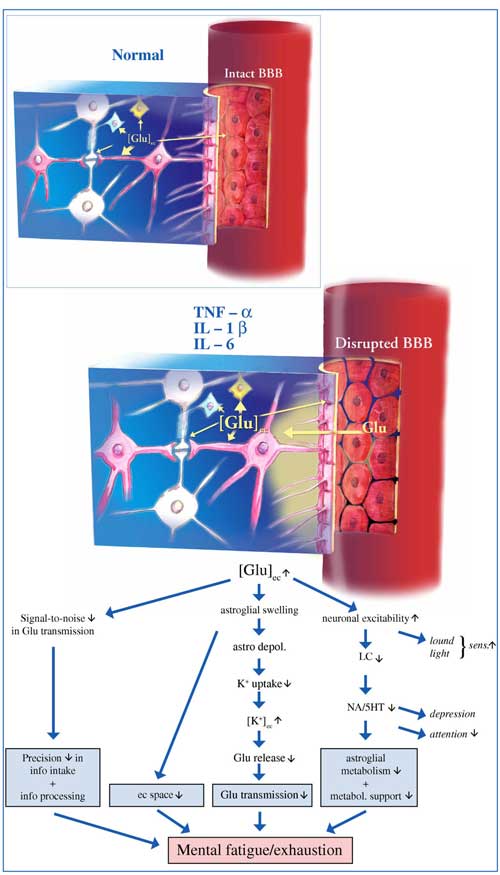
Studies suggest the immune findings in ME/CFS could explain the glutamate issues Marco believes could explain mental fatigue and exhaustion present in this disorder
The work of Julia Newton’s team at Newcastle university examining fatigue and autonomic dysfunction in ME/CFS and the autoimmune disease primary biliary cirrhosis has been discussed here recently. Regarding fatigue in primary biliary cirrhosis Medscape states :
“The etiology of fatigue is unclear; although some evidence suggests that abnormalities of the hypothalamic-pituitary-adrenal axis, decreased release of serotonin, and increased production of proinflammatory cytokines (ie, interleukin-1 [IL-1], interleukin-6 [IL-6], tumor necrosis factor-α [TNF-α] ) may be responsible.”Medscape
Intriguingly, a just published paper (Raszeja-Wyszomirska et al, 2013) reports the association between polymorphisms in TRAF1-C5 – a gene regulating TNF-a with ‘sickness behaviour’ – fatigue, mental and physical function and mood in patients with primary biliary cirrhosis. This finding appears to parallel increased polymorphisms in a TNF-a promoter gene in ME/CFS patients (Carlo-Stella et al, 2006) which led the researchers to comment :
“We hypothesize that CFS patients can have a genetic predisposition to an immunomodulatory response of an inflammatory nature probably secondary to one or more environmental insults of unknown nature.”
Collectively then, the consistently noted immune disturbances found in ME/CFS all accord with the notion of a neuroinflammatory state.
NK cell cytotoxicity
Oxidative stress alone, in conditions such as type II diabetes and renal failure (Berrou et al, 2013) may also account for reduced NK cell cytotoxicity through dysfunction or apoptosis of NK cells (Betten et al, 2004; Wang et al, 2009).
However one NK cell finding requires further explanation. Brenu (Brenu et al, 2012) reported that “significantly lower numbers of CD56brightCD16- NK cells” may lead to a greater susceptibility to viral and other infections and may be a ‘biomarker’.
CD56(bright) NK cells make up a minority of NK cell types but represent the majority in lymphatic tissues and are associated with sites of inflammation (Chan et al, 2007). While the exact proportion of these cells varies, the percentages are expanded or reduced in a certain number of diseases including coronary heart disease, allergic rhinitis and juvenile rheumatoid arthritis (Brenu et al, 2012). While, the significance of these variations is not yet clear (Poli et al, 2009), their presence does suggest that ME/CFS fits in a broad realm of inflammatory disorders.
Viral Persistence
Most viral studies in ME/CFS were done years ago and are dated and according to Bansal et al. the results are not particularly promising. However they do note some evidence that does suggest an intriguing herpesvirus/natural killer cell interaction :
“While the prevalence of positive serology for the common herpes viruses appears no different from healthy controls, there is some evidence of viral persistence and inadequate containment of viral replication. The ability of certain herpes viruses to impair the development of T cell memory NK cells may explain this viral persistence and the continuation of symptoms.”
Again viral persistence is consistent with impaired NK cell function and is discussed further below.
ETIOLOGY/ONSET
Similarly to the notion of ‘heterogeneity’ of symptoms, the pattern of onset reported by patients is also heterogeneous.
Viral/Bacterial

Pathogens and the role they play have been a question mark since the beginning
The various ‘epidemic ME’ clusters cast a long shadow over ME/CFS research (and how we conceive ME/CFS) and, as in the recent case of XMRV, a single pathogen model may initially appear attractive. Numerically, though, ‘epidemic’ cases must represent only a tiny proportion of those diagnosed with ME/CFS and research has shown that only a small proportion of people infected with common viruses will develop ME/CFS regardless of the pathogen, suggesting it is the host response to infection which matters. (Cameron et al, 2006; Katz, Jason, 2012)
Nonetheless, many (but far from all) people with ME/CFS report an acute infection immediately prior to the onset of ME/CFS indicating that viral (or bacterial) infections are a common trigger. As previously seen in Part III, various infections have also been seen to apparently trigger diabetes, stiff person syndrome and anti-NMDA receptor encephalitis.
The Japanese encephalitis virus (JEV) could provide something of a model for ME/CFS. This virus has been shown to cause encephalitis via a mechanism whereby TNF-alpha inhibits glutamate re-uptake leading to raised extracellular glutamate and excitotoxicity (Chen et al, 2012) :
“Our findings support a potential link between neuroinflammation and the development of excitotoxic neuronal injury in Japanese encephalitis. The link between neuroinflammation and excitotoxic death may involve a mechanism in which TNF-α released by microglia plays a facilitory role in glutamate excitoneurotoxicity via up-regulation of glutamate synthesis and down-regulation of glutamate uptake.”
Note that this process is remarkably similar to that proposed by Rönnbäck and Hansson to cause mental fatigue.
Psychological stress
We also saw in Part I that ‘psychological’ stress (isolation rearing) can induce a sensory gating deficit in rats and that (as discussed in Part II) this deficit may be a measurable artefact of a glutamate/GABA imbalance. Many people with ME/CFS report a period of elevated stress prior to or around onset, a ‘correlation’ that led some to conclude that ME/CFS is largely a ‘mood’ or ‘stress’ related disorder. Others, however, report that there were no unusual feelings of stress or stressful events associated with onset.
As discussed above, both acute traumatic stress and chronic stress may cause lasting glutamate induced dysregulation of the HPA axis. We also saw in Part I that the dopamine and norepinephrine (and indirectly glutamate) modulating COMT gene has been associated with ME/CFS in three studies. The low activity met allele of COMT is associated with increased pain sensitivity, executive function that deteriorates under stress and may mediate the emotional response to negative stimuli and stress (Smolka et al, 2004).
Prevalence estimates for the met/met allele equate to approximately 25-30% of the European population (SNPedia) with the low activity met polymorphism more common in women (Tunbridge, 2010 – Press release). It may be that psychological stress represents a more significant stressor for people who have one or more met alleles of COMT.
Strenuous exercise
Exercise may be another stressor that ‘kindles’ the vicious neuroinflammatory cycle. Many patients report having been very physically active prior to onset and a large scale prospective study (Harvey et al, 2008) suggests that those who exercised frequently are at greater risk of developing ME/CFS. Strenuous exercise of course also induces oxidative stress and high demands on mitochondria and this could be an initiating factor in the ‘vicious cycle’. As discussed above, ME/CFS patients appear to have an attenuated response from the normally protective heat shock proteins.
Autoimmunity

PANDAS is an autoimmune ‘neuropsychiatric’ disorder associated with an bacterial infection that sometimes responds to immune drugs
In the context of a possible autoimmune etiology of ME/CFS discussed in Part II, it’s notable that CD56 bright NK cell counts discussed above may be used to determine how effective the monoclonal antibody daclizumab is in the treatment of relapsing/remitting multiple sclerosis (RRMS). This supports the immunomodulatory role natural killer cells play in reducing brain inflammation and slowing disease progression in RRMS. (Bielekova et al, 2006) :
“…daclizumab therapy was associated with a gradual decline in circulating CD4+ and CD8+ T cells and significant expansion of CD56 bright natural killer (NK) cells in vivo, and this effect correlated highly with the treatment response”
This suggests that the low levels of CD56 bright natural killer (NK) cells seen in ME/CFS could reflect similar ‘brain inflammation’ and a possible autoimmune etiology.
As seen in Parts II and III, a number of conditions which involve a glutamate/GABA imbalance also now appear to have an autoimmune component including stiff person syndrome, anti-NMDA receptor encephalitis, late onset autism, autoimmune encephalitis and potentially type II diabetes, fibromyalgia and schizophrenia/schizoaffective disorders. As discussed above primary biliary cirrhosis is an autoimmune disease sharing many symptoms with ME/CFS where TNF-a may contribute to neuroinflammation.
An endogenous retrovirus?
Having apparently got one retrovirus out of our systems (pardon the pun), the results of Brigette Huber’s research on the involvement of the endogenous retrovirus HERV-K18 in ME/CFS have been long-awaited.
Human endogenous retroviruses (HERV’s) are the legacy of previous, often ancient, encounters with retroviruses which have become part of the human genome. While previously considered to be ‘inactive’ they can be reactivated by various environmental influences (including common pathogens) and are now implicated in the pathology of a range of diseases including cancers, nervous system diseases and autoimmune rheumatic and connective tissue diseases (Balada et al , 2009). The (not exhaustive) list includes rheumatoid arthritis , Systemic Lupus Erythematosus (SLE), Sjögren’s syndrome (Balada et al , 2009), multiple sclerosis (Laska et al, 2012), schizophrenia and bipolar disorder (Diem et al, 2012).
HERVs and ME/CFS
Similarities in the symptoms between ME/CFS and certain autoimmune diseases such as multiple sclerosis and Systemic Lupus Erythematosus (where HERVs may play a role) have led some investigators to look for and find a putative association between HERVs and CFIDS/ME/CFS (Urnowitz and Murphy, 1991; Huber – Conference press release, 2008).
Huber has previously presented data that suggests that HERV K18 may be reactivated in some ME/CFS and multiple sclerosis patients and that this reactivation may result from an initial infection by Human Herpes Virus -6 or Epstein Barr virus. These preliminary findings appear not to have been replicated in a more stringent study that failed to find evidence of higher levels of either HHV-6 or HERVs in the blood or saliva of ME/CFS patients (Oakes et al, 2013) :
“We fail to demonstrate a difference in HERV-K18 env transcripts, HHV-6 viral copy number, and HHV-7 viral copy number between CFS patients and healthy controls. Our data do not support the hypothesis of reactivation of HHV-6 or HHV-7 in CFS.”
Notwithstanding this negative finding, HERV’s have been implicated in a number of the neuroinflammatory conditions discussed previously and recently antibodies to a range of HERVs (including HERV K) have been detected in the tissues of the gut in a small number of ‘ME’ patients but not in controls (Lombardi et al, 2013).
HERV involvement in human diseases and the specific mechanisms involved is too vast for this review however, in the context of the current discussion, I’ll make just a few observations.
HERVs in ‘Neuroinflammatory’ diseases
Variants of HERV-W and HERV-K have been associated with schizophrenia and bipolar disorder (Diem et al, 2012) and may be reactivated by common pathogens including influenza virus, herpesvirus and Toxoplasma gondii (Perron et al, 2012) :
“HERV-W envelope gene (env) is activated by environmental factors and encodes a protein displaying inflammation and neurotoxicity”
“The seroprevalence for Toxoplasma gondii yielded low but significant associations with HERV-W transcriptional level in a subgroup of BD and SZ (bipolar disorder and schizophrenia), suggesting a potential role in particular patients.”
In the context of the association between ‘neuroinflammatory’ diseases and diabetes, polymorphisms in HERV K may be associated with a higher incidence of type II diabetes in schizophrenics (Dickerson et al, 2008; Nyegaard et al, 2012).
HERV impact on glutamate and GABA?
HERVs are known to ‘make use of’ many elements of the human biological machinery in order to replicate – including binding to the receptors of several neurotransmitters including the glutamate transporter ASCT2 receptor (Marin et al, 2003) lending some ‘construct validity’ to the role of HERVs in a ‘glutamate model’ of schizophrenia. Needless to say parking an endogenous retrovirus on top of neurotransmitter receptors might very well interfere with the normal functioning of those receptors. This ability has been hypothesized as being the result of the (ongoing) ‘arms race’ between viruses and the host immune system and enables HERVs to gain access to a wider range of human tissues.
One recent hypothesis (Hegyi, 2013) suggests that the long terminal region (which is known to silence, enhance or regulate gene activity) of HERV-W may downregulate the GABA receptor B1 creating a GABAergic deficit in schizophrenia. The author further suggests that the immune response to suppress HERV transcription may also suppress the activity of GABA.
While schizophrenia is obviously not ME/CFS it may share some sensory gating issues with it and other disorders including ME/CFS which might suggest common biological pathologies. Both of these findings offer a plausible mechanism for an association between HERVs and ‘neuroinflammatory’ conditions involving ‘mood’, sensory processing problems (and potentially movement disorders).
Antiretrovirals as a treatment for multiple sclerosis?
A clinical trial is currently planned to investigate the efficacy of an antiretroviral in multiple sclerosis (Giovannoni, Queen Mary University of London) where, as discussed above, CD56bright NK cell counts may act as a biomarker for the efficacy of daclizumab treatment :
“There is accumulating research evidence that Human Endogenous Retrovirus (HERV) and herpes viruses (in particular Epstein-Barr Virus) are involved in the pathogenesis of multiple sclerosis”.
People with active MS have higher levels of HERVs than people either without MS or who have other neurological conditions. It has been shown that HERVs may produce neurotoxic proteins/antigens associated with MS activity and disease progression.
This is the first clinical trial investigating the hypothesis that the antiretroviral drug raltegravir may suppress HERV activity and ameliorate progression of relapsing remitting MS.”
Gender
Significant gender imbalances are common in autoimmune disease (more females), autism spectrum disorder (ASD) (males) and ME/CFS (females). The protective effects of the estrogen mediated RORA gene may help explain the gender imbalance in ASD. Assuming that gender imbalances in ME/CFS are not merely an artifact (e.g. perhaps resulting from under-reporting or alternative diagnoses in men), as previously discussed, COMT gene polymorphisms that affect dopamine levels (and indirectly glutamate levels) could play a role in the gender imbalance in ME/CFS. As discussed in Part II recent research strongly suggests that gender (or more specifically the effects of estrogen) impacts on COMT functioning could lead to sexually dimorphic effects on brain function.
Estrogen also appears to have significant effects on some of the genes associated with ME/CFS – i.e. if you’re female, estrogen’s effects on the RORA gene may protect you against autism but if you have a certain form of the COMT gene, your brain may more vulnerable to ME/CFS.
Sudden Vs Gradual Onset – Were We Ever ‘Well’?
Once again, ME/CFS patients report different types of onsets with some reporting a sudden dramatic onset of symptoms (often following a viral illness) or a more gradual onset over a period of time. Some also report a ‘morphing’ of symptoms over time – often described in the literature as symptoms that are ‘protean’. In both cases most patients however report few prior physical complaints.
As discussed in Part III, infection appears to be able to trigger a number of conditions involving a glutamate/GABA imbalance and from this respect, if the neuroinflammatory cycle also underpins these cases, may represent a ‘de novo’ pathology.
However the ‘Dubbo’ and post-infective mononucleosis studies discussed above do also suggest that developing ME/CFS requires a combination of a pathogen and predisposing factors.
My introductory biography did state that I was fully physically healthy prior to the onset of ME/CFS. This is true, but it is also an error of omission in that I didn’t state that I had always experienced a high level of anxiety and an enhanced stress response plus some ‘characteristics’ (poor social skills, poor face recognition, sensory issues, delayed speech) that might suggest some ASD like tendencies.
I would propose that at least some ME/CFS patients might recognise similar characteristics prior to the onset of ‘physical illness’ but and that some of these symptoms may have increased post onset (others may have developed some of these characteristics afterwards and others may not have them at all). I suggest that these ‘atypical’ symptoms may be early indications of a predisposition to developing this neuroinflammatory cycle.
A one, two or multiple ‘hits’ etiology
More than likely all of the above factors act as stressors individually or in concert which may overwhelm a system that is predisposed to illness of various forms via the development of a neuroinflammatory vicious cycle. This predisposition may be genetic and could involve antioxidant, immune, mitochondrial integrity, neurotransmitter balance or an autoimmune or perhaps even a HERV component.
In this respect the model proposed is similar to that set out by Beatrice Golomb for Gulf War Illness and overlapping conditions such as CFS and ASD where a wide range of putative stressors in GWI may all result in the same pathological process involving oxidative stress/mitochondrial dysfunction with many downstream effects including a vulnerability to autoimmunity (Golomb, 2012).



Thank you for all of the wonderful info. Have been waiting for 30 somewhat years
Marco,
I appreciated this article. Since this is something that I have felt may be playing a role in POTS and have been doing research on it myself. I find the symptoms/presentations of POTS and ME/CFS to be so similar – yet different. There must be a connection, as some people have both.
One thing of note – from what I can gather from my reading, there can also be a mutation in the BHMT pathways of the methylation cycle that can mimic a COMT mutation. Especially an issue with BHMT4, can act the same and cause similar issues with glutamate function. It also can cause too much ammonia and using Yucca can help to neutralize that and helps in keeping the gut flora in balance and in breaking down proteins properly so that ammonia is eliminated. It is also suggested that B12 will help in the proper conversions necessary. This is explained in an article by Dr. Mullan and Yasko, it’s entitled “Gastrointestional Balance and neurotransmitter formation”. http://www.scribd.com/doc/98433639/Gastrointestinal-Balance-and-Neurotransmitter-Formation
Interestingly, in regards to myself – it was found that I have a protozoa that is very similar to malaria (FL1953 – Protomyxzoa Rheumatica), and I also had a co-infection that goes along with Lyme disease. Working on breaking down the bio-films that can prevent the immune system from recognizing these organisms and virus, bacteria etc. and eliminating things that could affect their flourishing, along with anti-malarial herbs and low dose antibiotics – has been of great help to me. I think it will be a long slow process – but, I’m considerably better. There is a close connection between the protocol for this and the mutations that I found in my methylation pathways. That connection may also exist in the connections with ME/CFS and POTS. Could it all be the same issues? Why might one persons immune system work properly and they never get sick, even from the same exposures. While another person gets sick and can’t seem to pull themselves out of it. Could these mutations in our genetics be causing these issues? I think time will tell.
Thanks for this article. It has given me a little more to think about and research.
Issie
Thanks Issie
That’s an interesting article.
Gastro issues (upper and IBS) have been a constant problem for me. A full work up showed ‘idiopathic’ malabsorption (no treatment recommended) and I was on Tagamet and Zantac for years until the upper gastro symptoms just disappeared.
I was tested for and clear of H-pylori.
If I could sort my gut issues out I feel there would be major improvements in other areas too. But where to start without positive tests?
The thing is, prior to ME/CFS I had a pretty cast iron stomach but I still had issues with anxiety etc which suggests a predisposition to develop systemic inflammation.
Inherited or congenital ‘weaknesses’ plus ‘stressors’ would seem to fit my particular case.
One of the best things I’ve done for myself is go low-fat vegan. It has helped my digestion, elimination and assimilation so much. I now have moons in my fingernails – showing assimilation of nutrients. Not only has it helped my intestinal issues – it also addresses the immune system. This is one thing that we can do without any type of testing. The hardest part is deciding to do it and then committing to it. Something wonderful that my diet has done is to reverse Chronic Kidney Disease – stage 3 to stage 1. That is unheard of. Most docs will say once your kidneys are damaged there is no redemption. But, my diet has done this.
Since I have MCAS (mast cell activation syndrome) I also use Allegra and Zantac – but, in much lower amounts than most people with this. One of the best meds I’ve used is GastroCrom. It also helps with the immune system. (I wouldn’t be on it without insurance – it’s unreal expensive.)
Many of us have found with 23&me testing, genetic mutations that we can work on and maybe prevent future issues with them. It is not a very expensive test. But, will require a whole lot of research on your part to interpret and figure out what to do with the data. This has been very beneficial to me. There is so much to learn about the body and how it works. It’s very intriguing to me and makes me realize how wonderfully made our “Creator” made us. Genetics and those mutations could play a big role in what “illness” we deal with and also in how we eliminate and process supplements, drugs, etc. Having a better picture of our own individual blueprint – can make a big difference in our approach.
There is a lot we CAN do. We just have to commit to doing them. That’s the hard part. There is no “magic pill”. If we do what we have in our own control and see where that takes us —then maybe doctors can address what is left over. It is very liberating to take over that control – knowing we are doing the best we can for our bodies. I do believe that genetics play a big role with us all. And working with that and the immune system and inflammation may be the best things we could do for ourselves. It has made AMAZING changes for me!
Issie
‘Self-help’ approaches are the only things that have had any benefits for me Issie, often discovered by chance.
Realistically speaking also, even if the science identified the core problem tomorrow there would be a long lead time even for drug repurposing and new drug development could be 15 years away – assuming the money and will was there.
Many of us don’t have much of a life to look forward to 15 years from now.
What the emerging science can do though is give us pointers to better target our own efforts.
In the context of the proposed glutamate/GABA imbalance I really should try to reduce my meat intake – it wouldn’t be hard as my wife is vegetarian. I’m just such a committed carnivore and I do feel that I need the protein.
It might be worth a try though – just to see if my digestion improves.At the moment lunch (usually light) if immediately followed by bloating and a major slump in energy such that I usually need an hour or two sleep afterwards.
Give it a try and just see if it makes a difference. You have to make up your mind to commit. That’s the hard part and then once your committed, doing it is the easy part. It was an absolute necessity for me and I’m so glad that I embraced it. Keep us posted.
Issie
I think it is time that every study reports the status of noradrenaline levels, cortisol, Glutahione, NK cel function, TNFa and HRV in there CVS population. These findings are consistent.
I’m inclined to agree Gijs.
There do appear to a limited number of consistent number of objective abnormalities including certain pro-inflammatory cytokines, reduced NK function and ANS dysfunction that should point to the underlying pathology even if we can’t nail it down causally to a certain pathogen, gut issues or whatever. I’d also like to see neurotransmitters such as glutamate and GABA measured but blood measures don’t necessarily equate to what’s happening in the brain and levels may vary by brain area.
But we do know many of the detrimental effects of high TNF-a. If so can it be treated (Fluge and Mella are currently trying Etanercept) and what effect would this have on the symptom burden – even without knowing exactly why they are raised.
I think the problem is that these findings are far from specific to ME/CFS. That’s a problem if you’re looking for a biomarker and want to demonstrate that you can distinguish X condition from Y. It’s not a problem however if you accept that the same pathological process may be common to many neurological conditions, merely manifesting in different symptom patterns.
It would be nice to be able to measure all these things – with science. But, with some things science isn’t there yet. We need to go with what our “gut” is telling us and work on what IS within our control. Things that we — with good sense — CAN do, this minute. For us, with scientific minds and wanting concrete proof of something, that may be hard. We want the science behind it. But, until that is available – maybe just addressing what we suspect – may make a huge difference in our “quality of life”. Hey, we’re all looking for that “purple Band-Aid”. It may be up to us to find it.
Issie
I’m trying to think of something I would add……
A citation page would be nice; there’s something I’d like to show my doctor and now I have to google it and hope I find it!!!!
Hi Marlee
You can find citations for Neuroinflammatory series here. . The citations for the last blog are at the bottom.
Fantastic articles!
I never post anything, but these were the most excitement in reading I have ever had in the ME world!
I thought I maybe could add a litlle.
Since I have a son with spastic diplegia (cerebral palsy) and relatives with Aspberger’s autism, ADHD etc, after reading the articles I did a little research and found the below quotes…
“In spastic diplegia (cerebral palsy) in humans, GABA absorption becomes impaired by nerves damaged from the condition’s upper motor neuron lesion, which leads to hypertonia of the muscles signaled by those nerves that can no longer absorb GABA.”
https://en.wikipedia.org/wiki/Gamma-Aminobutyric_acid
and from Pub Med:
“The current understanding of contributors to the risk for cerebral palsy is still incomplete. Multiple causes may interact by way of excitotoxic, oxidative, or other converging pathophysiologic pathways.” http://www.ncbi.nlm.nih.gov/pubmed/14596546
So here’s my thought:
I wonder now if cerebral palsy should now be added to the conditions to look for on the family tree of medical concerns, as another possible indicator of a familial pattern of GABA / Glutamate dysregulation.
This would address a medical issue (cerebral palsy) that I have never seen included in any lists of a family of conditions (with ME) also having no known etiology.
Thanks a lot Doug.
That’s really interesting. It sounds as if the hypertonia in cerebral palsy has similarities to stiff person syndrome.
Cerebral palsy didn’t turn up in my initial list of related conditions as there doesn’t seem to be any research indicating a sensory gating deficit which was the issue that suggested that ME/CFS may share a pathology with other neuroinflammatory conditions.
But a quick googling around suggests that those with cerebral palsy may also have sensory processing issues as well as motor problems. Have you by any chance recognised these sorts of issues with your son?
Re familial predisposition, the problem with family studies is that they look for increased incidence of one condition be it ME/CFS; fibro or whatever whereas I would suggest that they really need to look for increased incidence of a wide range of disorders that may be underpinned by the same neuroinflammatory pathology.
The same pathophysiology may show up in a family as a variety of seemingly separate and apparently unrelated conditions and conventional ‘diagnosis based’ surveys wouldn’t pick this up.
Hi Marco,
I should point out by way of a starting point, that the vast majority of cases of cerebral palsy have no know cause, no lesions, nothing remarkable (as of the last researching of this done by me years ago). I’m not a good researcher, and I don’t have the free access to follow up on this on scholastic databases as I would like.
I do know that my son had an operation called a “selective dorsal rhizotomy” operation (neuro-surgery on the sensory nerves of the dorsal root ganglia of the lower end of the spinal column).
This surgery was done, as it was explained to me 20 years ago, to address an over amplification by the brain of what were “normal sensory inputs” from an extremity of such as: proprioception (ability to sense the position and location and orientation and movement of the body and its parts), pain, temperature, or pressure etc.
Said another way, in C. P., the brain’s interpretation of certain normal sensory inputs (as seen when the surgeon’s milli amp signal is applied to the patient’s sensory nerves inter-operatively on the exposed spinal column during the surgery) elicit a motor response back to the legs that is many times what was “normal” (hence resulting in the “spasticity” that was immediately seen in those dysfunctional brain responses during the surgery).
In electrical systems we would call this an “excessive gain” in the circuit.
In a brain however, it seems we can’t get away from the aspect of “the perception” by the brain of the signals being provided to it…
… and this perception could very well have something to do with the a hyper-arousal state of the brain’s interpretive circuits.
It would be more than interesting to see family medical histories of CP patients regarding the:
1. occurrence of the medical conditions that also currently have no know etiologgy (e.g. MS autoimmune, autism ADHD etc. that you so well list)
2. occurrence of aberrant genetic SNPs regarding the methylation cycle etc
… to get a reasonable and scientific explanation of cerebral palsy, other than the “hypoxia” theory.
The hypoxia theory let’s remember, has had it’s application in the MECFS world as well (see Dr, Bell’s book “Cellular Hypoxia” 2007 – a book I would love to edit if it were to be released again…
see: http://www.goodreads.com/book/show/1589451.Cellular_Hypoxia_And_Neuro_Immune_Fatigue
Hi again Doug
I must say that even with the little I read the hypoxia explanation did seem a little ‘pat’.
The dorsal root ganglia do seem to keep cropping up in ME/CFS.
Interesting that you talk about excessive gain. My earlier articles on sensory gating suggested that the problem may be one of a reduced signal to noise ratio and that the constant bombardment of sensory input could contribute to a self perpetuating neuroinflammatory cycle. But excessive gain, as you know, also has a tendency to result in feedback loops especially if, in the brain, clearance of glutamate or inhibition by GABA is impaired which would normally attenuate these signals.
Hypoxia, Ischemia and energy starvation (see Cort’s recent blog : http://www.cortjohnson.org/blog/2013/07/01/your-brain-without-energy-broken-brain-energy-loop-wipes-out-cognitive-functioning-gws-me-cfs/) all appear to result in the same neurotoxic state, as does thiamine deficiency (see Cort’s other recent blog : http://www.cortjohnson.org/blog/2013/07/05/is-simple-relief-from-fibromyalgia-mecfs-found-early-reports-spark-interest/) which interferes with astrocytic transporters that usually clear extracellular glutamate.
Thiamine supplementation and N-acetycysteine (NAC) may help prevent Wernicke’s encephalopathy in alcoholics :
http://www.ncbi.nlm.nih.gov/pubmed/19565658
Interestingly NAC is one substance I keep coming across (just taken some actually to help deal with heat intolerance) and has been proposed as potentially helpful (presumably within a window of opportunity) in cerebral palsy :
http://www.ncbi.nlm.nih.gov/pubmed/22517883
So many overlaps in ‘medically unexplained’ conditions.
LDN seems to help with “auto-immune” conditions and works very well for some people with ME/CFS it is inexpensive and worth a try at least. Not compatible with opiate drugs for pain but otherwise very few side effects once the body gets used to it. Best to start low and go slow. Works for me!