

Day two of the Conference – not all of which is covered here, started off with a physicians’ panel, then NIH Director Francis Collins showed up (covered later) just before the Intramural study presented some results, then it was onto Ian Lipkin, Dr. Oh, Bhupesh Prusty and Ron Davis.
Physicians Panel
The ME/CFS experts on the panel spent 10 minutes on a topic of their choosing. Dr Montoya reported that some of his long term patients with herpes viral infections were doing quite well on antivirals. Dr. Levine covered mast cell activation in 10 minutes – a valiant effort! I didn’t cover it but if you’re interested check a blog which includes a longer presentation at the 2018 Dysautonomia Conference.
Dr. Bateman – Orthostatic Intolerance
“Orthostatic intolerance is the low-hanging fruit. It is diagnosable and treatable, and we have a lot of literature about how to deal with it.” Dr. Bateman
Dr. Bateman spent her ten minutes on orthostatic intolerance – the inability to stand or stay standing without symptoms flaring up.
 She, like others have, warned how quickly bed rest can cause orthostatic intolerance and had some clever ideas about how to exercise as safely as possible.
She, like others have, warned how quickly bed rest can cause orthostatic intolerance and had some clever ideas about how to exercise as safely as possible.
 Health Rising’s Quickie Summer Donation Drive is On!
Health Rising’s Quickie Summer Donation Drive is On!She has added pyridostigmine bromide (Mestinon) to her bag of tricks for fighting orthostatic intolerance (OI). Mestinon is another helpful drug which has been hiding out in plain sight for decades. Brought to the attention of the ME/CFS community by David Systrom, it provides another option for dealing with OI and energy issues.
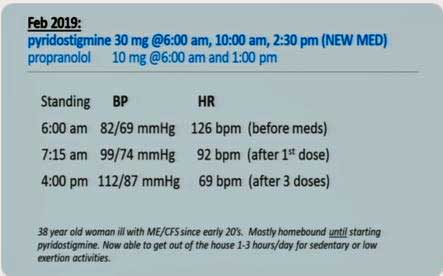
Mestinon completely cleared up one person’s POTS. (Not everyone has such a dramatic result.)
Check out a superb overview on Medscape by Miriam Tucker which goes over Dr. Bateman’s talk and David Systrom’s findings on exercise intolerance in ME/CFS. In it ,Tucker reported that the rationale for using Mestinon is to enhance norepinephrine release at the postganglionic synapse, thereby improving the constriction of the veins at the site of exercising muscles. It appears Systrom believes that impaired blood flow from the muscles is reducing blood flow to the heart. Increasing blood flow from the muscles improves blood flow to the heart, and ultimately better flow of oxygen to the muscles. One excellent Mestinon ME/CFS responder was able to exercise for the first time in decades.
Rowe reported to Medscape that Mestinon can be “transformative for some patients… even if they have not responded to fludrocortisone, midodrine, or beta-blockers”, and Dr. Bateman stated that Mestinon has “been an interesting and very effective intervention” and relayed the case of one responder.
Dr. Bateman also uses midrodrine, low dose propanolol, compression stockings, hydration and others in OI. Check out her video below.
- Check out home tests and treatment options for OI in Health Rising’s Orthostatic Intolerance Resource section.
Dr. Levine gave an outline of mast cell activation syndrome (MCAS) – a thankless task – in 10 minutes. Dr. Montoya reported that increased herpesvirus activation is found in his severely ill patient cohort (just the opposite of what Ron Davis found in his cohort) and that extended (as in over a year) antiviral herpes treatments do return some patients to health. Then, Dr. Peterson reported on the role of precision medicine in ME/CFS.
Dr. Peterson’s Precision Medicine Hopes
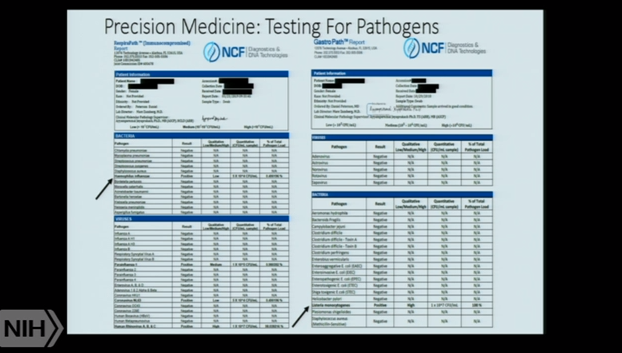 Dr. Peterson’s talk, which focused on the ever evolving world of diagnostics, was fascinating. He rattled off several cases of obscure diagnoses (Powassan virus, overt (and correctable) cytokine abnormalities, and others) enabled by technical advances in medicine. I was in contact with one person who Dr. Peterson diagnosed with uranium poisoning. Peterson is using tools like Genecept and the Precision Medicine Test for Pathogens to get ever more precise diagnoses.
Dr. Peterson’s talk, which focused on the ever evolving world of diagnostics, was fascinating. He rattled off several cases of obscure diagnoses (Powassan virus, overt (and correctable) cytokine abnormalities, and others) enabled by technical advances in medicine. I was in contact with one person who Dr. Peterson diagnosed with uranium poisoning. Peterson is using tools like Genecept and the Precision Medicine Test for Pathogens to get ever more precise diagnoses.
Plus, Peterson provided some more hope when he reported that a heart disease drug (of all things) called spironolactone may provide a new class of drugs to fight herpesviruses.
He and the Simmaron Foundation are also mining his database of patients to learn what works best in which patients, and the Simmaron Research Foundation is doing a deep dig into the effects of Ampligen. Results from that study were presented at the Young/Early Career Investigators’ workshop and will be presented on this site soon.
NIH Intramural Study Scores Some Early Hits
The fact that the Intramural study was able to deliver so little at a conference that was intended to show its results off suggests that everything is taking longer than expected. That seems to be a theme with the NIH and ME/CFS.
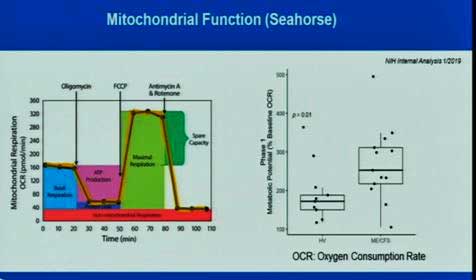 The good news is the intramural study is starting to do some preliminary analyses, and they are finding some things. Using – what else – the Seahorse, they’re finding increased (not decreased) mitochondrial respiration in the ME/CFS patients tested so far. This isn’t as far-fetched as it sounds. A Stanford study found that immune cells from ME/CFS patients had double the rate of energy production compared to cells of healthy controls. The study findings suggested increased glycolysis was the culprit. Increased folding of mitochondrial membranes called “cristae” (which results in increased membrane surface area) suggested that the mitochondria, even though they were pumping away, were under energy stress.
The good news is the intramural study is starting to do some preliminary analyses, and they are finding some things. Using – what else – the Seahorse, they’re finding increased (not decreased) mitochondrial respiration in the ME/CFS patients tested so far. This isn’t as far-fetched as it sounds. A Stanford study found that immune cells from ME/CFS patients had double the rate of energy production compared to cells of healthy controls. The study findings suggested increased glycolysis was the culprit. Increased folding of mitochondrial membranes called “cristae” (which results in increased membrane surface area) suggested that the mitochondria, even though they were pumping away, were under energy stress.
The author of that study suggested that immune activation could be driving the high energy production in the cells. Note, though, that these cells are being measured outside the bloodstream. Ron Davis’s tests suggest that something in the blood is tamping down the energy production of immune cells.
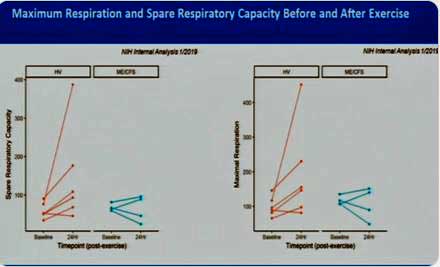
Exercise causes a drop in oxygen consumption (energy production) in about half of ME/CFS patients.
Where the intramural study really shines is in its use of a maximal exercise stressor to really shake things up. Much less data on this is available, but Walitt did show that the bout of exercise caused mitochondrial oxygen consumption to increase in the healthy controls but decrease in half of the ME/CFS patients (in blue) tested thus far.
The NIH is doing something else that hasn’t been done before – flow cytometry on cerebral spinal fluid from lumbar punctures and the blood at the same time. Walitt then highlighted another strength of the intramural study – the huge database of findings that NINDS has for other diseases that can be matched with ME/CFS – giving us, hopefully, some idea of what diseases ME/CFS is most closely allied with.
Among other things, flow cytometry allows researchers to minutely characterize the types of cells found in a sample. Finding elevated numbers of cells that get activated when a pathogen is present, or are indicative of an autoimmune process, gives researchers an idea of what’s going on. Finding low levels of cells could suggest a hole in our immune defense.
The NIH’s plan is to start off with a coarser filter and then dig down further to better characterize any anomalies they find.
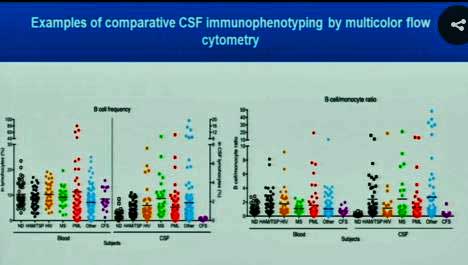
The very low purple levels on the right side of each diagram indicate a “bottoming out’ of B-cells in the spinal fluid of the ME/CFS patients.
The NIH found no difference, thus far, between CD4/CD8 ratios or NK cell frequency in ME/CFS and healthy controls, but check out what happened when they examined B-cell frequency: normal in the blood but very low – so low that Walitt called it a “bottoming out” – in the spinal fluid. This was particularly unusual, as it looked like every other disease in Walitt’s chart demonstrated increased B-cell frequencies. Another B-cell was similarly low in the cerebral spinal fluid (right diagram). This is another piece of evidence suggesting that the immune system in the brain may be a key player in ME/CFS.
The study protocol has been – as Nath suggested it would – shifting. (It’s now on its 11th amendment.) It appears that the NIH is now inviting participants who washed out after the first study to come back and do more testing (rTMS, fMRI, EEG, muscle and skin biopsies). These studies are now going to be included in the first week for all participants. One wonders if the NIH is finding these tests to be particularly discriminative.
A Colorado doctor who has a patient in the study got up and called the study an “unbelievable opportunity” and asked them to increase the number of participants.
This trial is taking time but is off to a good start with some positive preliminary findings. It needs more study participants! They would particularly love to have ME/CFS patients who have a healthy family member who can participate. Find out how to participate in this study here.
- Check out an interview with Dr. Nath on the Intramural Study
Ian Lipkin’s Possibilities
Lipkin first presented a map showing an impressive array of collaborators stretching from coast to coast. He’s all about rigor – and highlighted that fact on his slide. One gets the sense that Lipkin is not going to get ahead of the science – and he pointed out an important theme – large-scale studies, done correctly, start to add up.
 ME/CFS is, of course, full of small studies with interesting results that have done little to spark the imagination of other researchers. Large-scale studies done well that come up with consistent results is another animal entirely – and the NIH funded research centers, with their large studies, are helping with that.
ME/CFS is, of course, full of small studies with interesting results that have done little to spark the imagination of other researchers. Large-scale studies done well that come up with consistent results is another animal entirely – and the NIH funded research centers, with their large studies, are helping with that.
The three projects underway in the Lipkin lab demonstrate just how important these centers are. No other institution has the cash to fund, on an ongoing basis, three large projects like those underway at Lipkin’s center. For instance, the full genome analysis of the microbiome, underway at Lipkin’s NIH center, contains 195 cases and controls.

Lipkin finds consistent upregulation of harmful bacterial species and downregulation of helpful bacterial species in ME/CFS.
Lipkin’s now got two big microbiome studies under his belt. (The other one, funded by the Hitchens Foundation, has 100 cases and controls.) The good news is that these fantastically complex studies have come up with some similar results. Two bacteria associated with inflammation and gut permeability are upregulated in ME/CFS, while butyrate-producing bacteria which tend to dampen inflammation are reduced in ME/CFS.
Lipkin has found dramatically different gut bacteria in ME/CFS patients with IBS. Next, he noted that butyrate species were also reduced in this group and that two studies have found increased numbers of Clostridium species. The Clostridium bacterial group contains some notably pathogenic species.
The fact that Lipkin also found harmful bacteria in the saliva of people with ME/CFS associated with oral and gut issues made one wonder if any part of the microbiome is not “off” in ME/CFS.
His search for a virus – which he had great hopes for – has been for naught, however. A more detailed search for herpesviruses in white blood cells, saliva and even in fecal material has found nothing statistically significant in ME/CFS. Ron Davis, with his severe ME/CFS group, and the SMCI, with a previous study, have reported similar results.
That presents the odd paradox of knowing that some patients benefit from antivirals, without any evidence, thus far, that viruses are present. Lipkin did hedge his results a bit by saying that, at the time and in the compartment they’ve sampled in, they’ve been able to find nothing that would jump out at them. He also said that he believes a herpesvirus trial in patients with the right test results makes sense “biologically”.
Lipkin next presented evidence suggesting that the metabolomics field is not yet on sturdy legs. An apparently dramatic change in the databases researchers rely on over just a few years made it “very difficult” to compare the results of his recent NIH study and his last one – which was published just last year (!). That probably didn’t put a happy face on a researcher focused on rigor and reproducibility, but the good news is that, after finessing the data to account for the changes, a similar set of metabolites popped out that have shown up in his studies and in past ME/CFS metabolomic studies.

Low plasmalogen levels – an early but intriguing finding from Lipkin.
A fascinating but early result involves plasmalogens – lipids that play important roles in the structure of cell membranes in the heart and brain. They’ve shown up in both of Lipkin’s metabolomic studies and are reduced in the brains of Alzheimer’s, Parkinson’s, multiple sclerosis and – this preliminary findings suggests – ME/CFS. They’re degraded by a compound called cytochrome C that is released by the mitochondria when they are under stress – including the oxidative stress that is common in ME/CFS.
The plasmalogen finding, if it holds up, could provide a nice link between mitochondrial issues, the oxidative stress found in the brain and elsewhere, the issues of premature aging, and the nervous system problems in ME/CFS.
Lipkin’s pilot epigenetics study was notable for what he did with it – he added a transcriptomic analysis his study.
Transcriptomic analyses give “a broad account of which cellular processes are active and which are dormant.” Lipkin was able to tie those two analyses together in a nice package to show that genes associated with iron uptake, not only did not appear to be turned off, but the transcriptomic analysis indicated that they were less active in real time. The differences were modest, but Lipkin noted that modest differences can add up if they show up repeatedly in a pathway – which may be happening in ME/CFS.
If they are, they could have quite an effect. Lipkin stated that, based on their “very early” data, they would “predict” lesions would be present at four parts of the iron intake pathway into the cell, which plays a role in two potentially very important aspects of ME/CFS: oxidative phosphorylation (ATP production) and oxidative stress.
All in all, Lipkin’s talk presented some fascinating possibilities. It’s still early days – we don’t know if these results will be relevant to ME/CFS – but they certainly showcased the way these large and complex ‘omics studies may be able to provide some answers.
Coming back to the pathogen issue, Lipkin reported on a “serochip” he’s developed in collaboration with Roche, which allows him to use antibodies to detect previously undetected infections. The chip, in his work with ticks, for instance, allowed him to detect several new tick-borne infections.
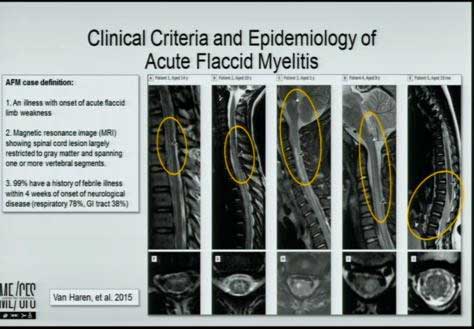
Virus studies were uninformative, but a new antibody approach worked.
He then referenced acute flaccid myelitis, a rare (1:2,000,00) but devastating neurological disease, which in its onset – a mild viral infection involving enteroviruses – has some similarities to ME/CFS. (Avindra Nath is also right in the thick of the hunt for the cause of the disease.) Identifying the pathogen producing the lesions in large sections of these childrens’ spinal cords has been torturous. Enteroviruses are highly suspected, but even Lipkin’s very sensitive techniques, done in the cerebral spinal fluid, have often proved fruitless.
A Serochip specifically designed to assess the disease, though, produced positive results in 87% of the children with the disease and not in other neurological disease. Lipkin, then, is on his way to possibly solving the greatest question in acute flaccid myelitis – is an enterovirus causing the disease? Lipkin’s results suggest yes, and can even point to the two enteroviruses likely to be the culprits.
His results suggest – as has been suspected in ME/CFS – that acute flaccid myelitis is a hit and run phenomenon: the pathogen strikes, sets a damaging process in motion, and then disappears. Lipkin’s success here suggests that if a putative trigger can be identified, then a specialized antibody panel can be produced to identify it. His next phase is to complete the fungal and other work (nobody to my knowledge has assessed fungi in ME/CFS yet) and then use a serological approach to dig deeper.
Lipkin’s talk, then, presented a nice blend of hard results – consistencies found in the microbiome and metabolomic studies – and intriguing possibilities which we’ll have to wait to see if they pan out.
Lipkin’s talk was another reminder of how good it is to have this researcher, with his wide array of sophisticated technologies, working on ME/CFS.
Dr. Oh – The Strain of Getting Gut Results
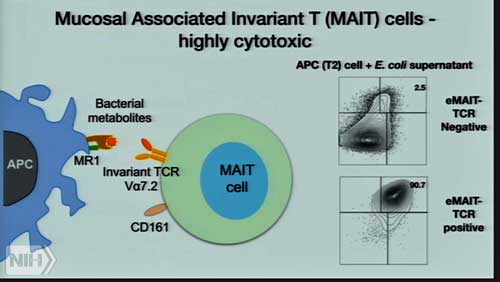
Are highly cytotoxic T-cells in the gut causing problems in ME/CFS?
Then it was on to Dr. Oh, who immediately shored up Lipkin’s gut findings with one of her own, indicating that butyrate bacteria are indeed reduced in ME/CFS. When she tried to add butyrate bacteria to ME/CFS cultures, they failed to thrive, suggesting that something in the teeming gut ecosystems of ME/CFS patients was knocking them out.
The question, of course, is what is turning up the heat (inflammation) in ME/CFS patients’ guts? The answer might be those MAIT T-cells Unutmaz referred to earlier – and the bad bacteria activating them.
 A search for bacterial triggers found so many different bacterial species that the sheer variety of them, Dr. Oh said, blew her mind. Then it got even more complicated. MAIT cells are known to be activated by antigens that are generated from a precursor of the riboflavin (Vitamin B2) biosynthesis pathway, but Oh reported that: (a) not all bacterial species that activate MAIT cells produce riboflavin; and (b) some that do produce riboflavin do not activate MAIT cells.
A search for bacterial triggers found so many different bacterial species that the sheer variety of them, Dr. Oh said, blew her mind. Then it got even more complicated. MAIT cells are known to be activated by antigens that are generated from a precursor of the riboflavin (Vitamin B2) biosynthesis pathway, but Oh reported that: (a) not all bacterial species that activate MAIT cells produce riboflavin; and (b) some that do produce riboflavin do not activate MAIT cells.
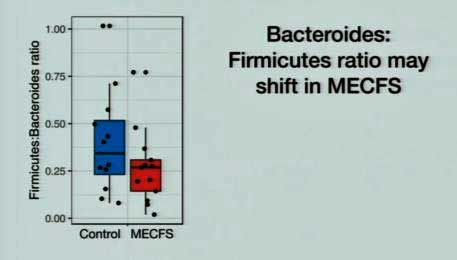
The fact that the reduced ratio of bacterioides/firmicutes bacteria found in ME/CFS is also found in other diseases such as inflammatory bowel disease and type 2 diabetes suggests it may be having negative effects.
When Oh said that it’s not actually “this easy”, I had to laugh as it didn’t seem easy at all, but then it did get a lot harder. Lipkin and Oh are looking at actual bacterial species – a big jump up in clarity for ME/CFS – but species, it turns out, can only take you so far. The real juice, Oh suggested, lies in the strains found within the species. How different can strains within a species be? Broccoli, cauliflower, brussel sprouts and cabbage are all strains of the same species. There are benign E. coli strains and deadly ones.
Precision gut therapeutics
So, the question becomes, which strains of bacteria best activate MAIT cells, and they are looking to find out. So far, they’ve found a huge difference in MAIT cell activation across different strains of a bacterial species.
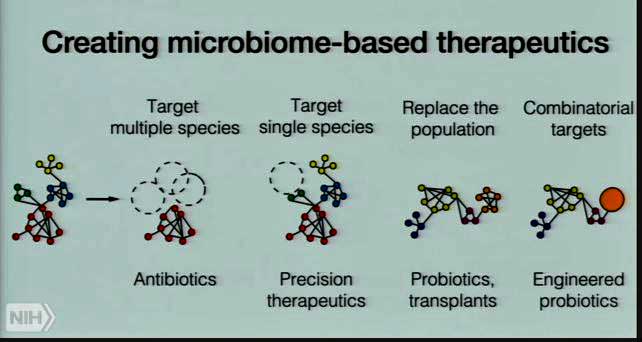
The goal in the end is precision microbiome therapeutics that seeks to return the microbiome to a healthy, non-inflammatory state. Antibiotics – long the preferred therapeutic modality – are too crude. Precision replacements, including probiotic transplants and even engineered probiotics are the wave of the future.
In response to a question about diet, Oh noted that they will include diet in their analyses. In fact everything – immune findings, gut findings, diet, etc. is all going into the big data mix. Diet does have an effect on the microbiome, and it may play a role in the therapeutic approach to ME/CFS. (It’s hard to believe that it could play a significant role in triggering it.) Oh noted that, as with our genes, we’re born with our own unique bacterial makeup, which diet can then affect to some extent.
Oh said it’s entirely possible that researchers will be able to create a specialized fecal transplant, which, through a single or a few bacterial species, addresses the gut issues in ME/CFS. When that might occur, she would not say…
Still, the Unumatz/Oh team has gone beyond the exploration phase; i.e., they have a pathway. They have a T-cell their research indicates may be a major player, they’re determining which bacterial species and even strains may be triggering the production of that T-cell, and if they can identify those, they’ll have a shot at turning MAIT cells down. If they are right, reducing the immune activation that so many people believe is driving this illness, may be the key.
Bhupesh Prusty’s Big Idea
“There is clearly something in the (ME/CFS) patient’s serum” Prusty

While exploring his big idea, Prusty came to the same conclusion as Ron Davis and Fluge and Mella: something in ME/CFS patients’ serum is affecting the ability of their cells to produce energy.
What an intriguing character Bhupesh Prusty is. Speaking just minutes after Ian Lipkin reported that he could find no evidence that herpesviruses are playing a role in ME/CFS, this young German researcher from the University of Wurtzburg suggested that HHV-6 infections are alive and well in ME/CFS and are putting a hammer to cellular energy production. The title of his talk “Pathogenic Alterations of Mitochondrial Dynamics: A Working Model for ME/CFS” said it all.
Funded by the SMCI’s Ramsay Awards to study ME/CFS (as well as the HHV-6 Foundation), he was one of the few researchers from outside the U.S. to present. The NIH must have thought he was doing something right to get him over here.
Over the past year or so, it’s become pretty clear that something is up with the energy production of our immune cells. Could it be a pathogen? In what was one of the more provocative talks of the conference, Prusty made it clear he thought so. The association between pathogens and ME/CFS is clear: from the Dubbo studies on, the data is very clear – whether it’s EBV or another herpesvirus, or giardia or Coxsackie or another pathogen, any serious infection is typically going to leave a subset of people in a long lasting ME/CFS-like state.
The big question dogging this field, of course, is how that happens. Prusty will present on HHV-6A data, but he believes that that pathogen, and herpesviruses in general, present the tip of the iceberg. He believes his model will also fit other pathogens.
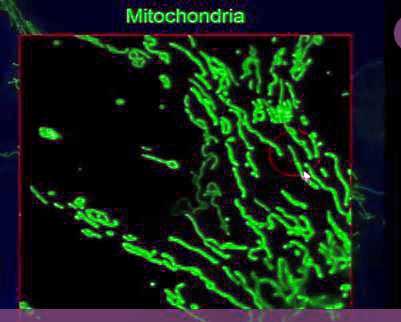
Not necessarily like the textbooks show at all…In their elongated state, the mitochondria play an important role in immunity.
Prusty, like Naviaux, believes the mitochondria are much more than the energy producers of our cells – they also play a key role in the cell’s antiviral response.
Prusty’s data indicates that HHV-6A – the more pathogenic of the HHV-6’s – prevents the mitochondria from combining together to form the elongated forms that participate in antiviral defense.
Prusty found that an miRNA was breaking the mitochondria up. HHV-6A not only promotes the expression of this miRNA, but produces a bit of RNA (sncRNA-U14) which also prevents the mitochondria from joining together to knock out viruses. Plus, viral reactivation of HHV-6 causes ATP levels to drop.

Suspect RNA is found throughout the herpesvirus family.
Prusty then showed that the piece of RNA he believes is jamming up the works of the mitochondria is found throughout the herpesvirus family. He believes he’s uncovered a “potentially universal” approach that pathogens take to affect the mitochondria.
But what about HHV-6A? Lipkin just reported that he was unable to find any evidence that HHV-6 is playing a significant role in ME/CFS. When Prusty looked and looked and looked he was unable to find much HHV-6 either. Even when the virus was present – the loads were low – not nearly enough high enough to cause something as severe as ME/CFS.
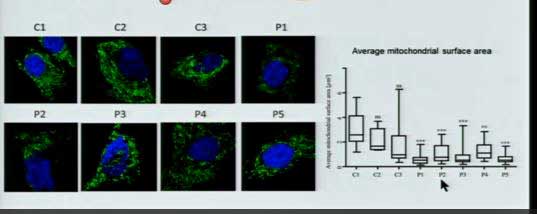
See the green (elongated mitochondria) disappear and the dip in the graph to the right as mitochondria surface area declines as the mitochondria begin to break up.
Prusty then looked for evidence that his mystery RNA was present in blood – and found it – in about 40% of patients and in none of the controls.
Then serendipity struck. Fluge and Mella had just published a paper, and Ron Davis had suggested that something in ME/CFS patients’ serum was interfering with ATP production – causing what Prusty called a “senescence” – something similar perhaps to the hypometabolism proposed by Naviaux.
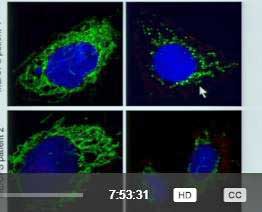
The mitochondria are in green. Again, adding ME/CFS patients’ serum (right hand pictures) into healthy cells causes the mitochondria to break up and decline.
When Prusty added ME/CFS patients’ serum to his culture, the mitochondria began to break up. Adding supernatant from an HHV-6 reactivation caused the same process to occur.
While only one study showing these findings has been published, the fact that serum from ME/CFS patients from the U.S., Norway and Germany appears to be producing the same results is good news indeed.
Referring to a graph which I couldn’t make sense of, Prusty reported that removing ME/CFS patients’ serum from the cells produced a startling transformation – the mitochondria began to come together and produce elongated forms. They appeared healthy again.
Prusty is attempting to isolate that mysterious, mitochondrial-inhibiting factor in an interesting place – the exosomes that Maureen Hanson is examining in her NIH research center. Exosomes – small vesicles that kind of burp free from the cells from time to time – are a different form of cellular communication – that is able to alter cellular functioning. If something in ME/CFS patients’ serum is causing problems, it could be an exosome. Alternatively, high levels of oxidative stress, small RNA’s or cytokines or other proteins could be the issue.
Prusty’s serum work set the stage nicely for Ron Davis’s presentation.
Ron Davis – Always Pushing Forward
Davis started his talk acknowledging that the title “Estabilishing New Mechanistic and Diagnostic Paradigms for ME/CFS” was a bit optimistic, but intentionally so because we always need to push forward.
He couldn’t be more correct. Our actions always align with the future we hold in front of us. If that future is one of resignation, of little change and progress, then our moods, our thoughts and our actions will correlate with that and things will probably go on as they have.

Big data indeed.
Resignation provides its own kind of juice. There’s a kind of righteous comfort in knowing that things will never change, that things are preordained to go the way they have. It gives you a sense of control, but it also takes you off the hook for trying to change things, and there’s ultimately no cheese down that tunnel.
If, on the other hand, we hold a future in front of us of this disease being validated and worked on, of sick people becoming well, then our thoughts, feelings and actions will correlate with that future and we will be freed up to move and take the actions that will ultimately bring those things about. I have no idea when those things will occur, but I do know that that’s a far more exciting and fulfilling future to live into. It even brings a sense of worthwhileness to the suffering.
The Gist
Orthostatiic intolerance is common, is often not diagnosed and often can be treated
Some of Dr. Montoya’s patients on longterm herpesvirus antivirals have responded well
Precision medicine is helping Dr Peterson uncover diagnoses that would have been missed before
Early results from the Intramural study suggest mitochondrial abnormalities and low B-cell frequencies in the spinal fluid are present
Three large gut studies have all found reductions of butyrate gut bacteria – suggesting that a pro-inflammatory gut ecosystem is present in ME/CFS. Two studies have found in increases in possibly pathogenic Clostridium spp
Both Ian Lipkin in his “normal” ME/CFS group and Ron Davis in his smaller severely ill ME/CFS group have failed to find any evidence of unusual viral activity. Lipkin did, however, note that specialized antibody panels prove fruitful in the quest to identify viral triggers in ME/CFS
The metabolomics findings continue to show good consistency across the studies.
Ian Lipkin’s early finding of increased plasmalogens could link oxidative stress, mitochondrial problems and neurodegenerative issues together.
Ian Lipkin’s very early epigenetics/transcriptomics results predict problems with cellular iron absorption and ultimately mitochondrial issues are present
Oh is searching for specific strains of bacteria that are triggering high levels of MAIT cells that Dervya Unutmaz believes may be causing major problems in ME/CFS.
Oh held out hope for a precision fecal transplant which returned the gut ecosystems of ME/CFS patients to normal.
Prusty, a young German research, finds that a small bit of HHV-6A RNA is able to stop the mitochondria from joining together to participate in antiviral defense. This RNA is found throughout the herpesvirus family.
in a rather stunning addition to the plasma findings from Ron Davis and Oystein Fluge, Prusty found that both the serum from ME/CFS patients and the serum associated with HHV-6 reactivation, caused the mitochondria to disassociate, thus turning off their antiviral defense.
Ron Davis is finding, as Jose Montoya did before him in less severely ill patients, that cytokine levels are correlated with severity in his severely ill cohort.
Assessing the validity of the Metabolic Trap hypothesis has hit a snag as neither filtering cells, changing the media, or improving the instrument used has resulted in results that can be trusted. Next, the group will assess the trap in individual dendritic cells.
The nanoneedle continues to be able to distinguish ME/CFS patients from healthy controls with an astonishing specificity and the results will soon be published in a distinguished journal.
Plasma exchange experiments continue to indicate that something in ME/CFS patients plasma is causing their cells to behave strangely when placed under (salt) stress. in a possible major breakthrough early testing suggest that exosomes – small vesicle cells emit which communicate with other cells – may be responsible. Both Maureen Hanson and Prusty ( and probably Ron Davis :)) are examining exosomes in ME/CFS.
Early hair analysis results highlight increased mercury and uranium (!) levels associated with decreased selenium levels. The sample size is small, however.
The point is, the future that Davis is living into – beating this disease as quickly as possible – requires collaboration and sharing data as broadly as possible. Given that future, Davis was able to find a way for Stanford to do something it had never done before – letting outsiders inside its gates.
It was good to see that cytokine levels do correlate with illness severity in Davis’s small but very severely ill ME/CFS group, as we’ve seen in Montoya’s much larger and less ill group. (Several times, Davis made clear that he believes his severely ill group is probably more severely ill than others; i.e. than Dr. Montoya’s!) This field appears to be developing a new way of looking at cytokines – which, after all the variable cytokine results, appears to work for ME/CFS. While introducing a new way of looking at the immune system bumps up against barriers, it seems it will be necessary in this category-defying illness.
The search for viruses, bacteria and parasites is not over, but it appears to be mostly over and nothing of consequence has shown up. An in-depth genomic analysis (provided for free by Illuminex) is still being assessed, but it did indicate that all 20 of the severely ill patients had damaging mutations in their IDO2 gene. Because those mutations are common, they would not have shown up in an automated search, but there was something different in the ME/CFS patients – many of them were homozygous; i.e. both alleles of their genes carried the mutation, rather than just one.
Davis described Robert Phair’s Metabolic Trap hypothesis, how high tryptophan levels can knock out the IDO1 enzyme, which in turn knocks out the serotonin pathway. There is a backup – the IDO2 enzyme – which converts the problem substance, tryptophan, to kyneurinine – thereby dropping tryptophan levels to the point at which IDO1 and the serotonin pathway are functioning again. Phair’s modeling work suggests that even a heterozygous mutation could so impair IDO2’s ability to drop tryptophan levels that the pathway could remain broken.
The thing about this trap is that, if it exists in ME/CFS (or other chronic illnesses), it would be very hard to get out of. In fact, Davis suggested that metabolic traps could explain why illnesses tend to be chronic and so difficult to get out of. Once you have one, you tend to have one for life. Phair and Davis have, in fact, uncovered another possible metabolic trap involving tyrosine.
The tryptophan trap model proposes that tryptophan (trp) will be increased, the kynurenine/trp ratio will be decreased, and the flux through that pathway will be impaired. The trap seemed set – the results of the first six patients accorded with it, but further testing revealed too much variability, indicating that the testing couldn’t be trusted. They went back, changed the media to boost the signal, switched to a better mass spectometer – and still got results that were all over the map. They filtered cells out to concentrate on the dendritic cells believed to have the trap – and are still getting too much variability. It may be, though, that the trap is present in only a subset of dendritic cells – so the next step is to examine isolated dendritic cells.
But this is how science goes! No one ever said it would be easy, and Davis has alluded numerous times to how hard it is and how complex the body is. Bumps in the road are expected.
Davis asserted that the Metabolic Trap hypothesis makes sense – it suggests, as Cortene’s hypothesis does (in a different manner) that serotonin problems are mucking things up in the brain. If it is present, the metabolic trap should affect the gut, brainstem and immune system.
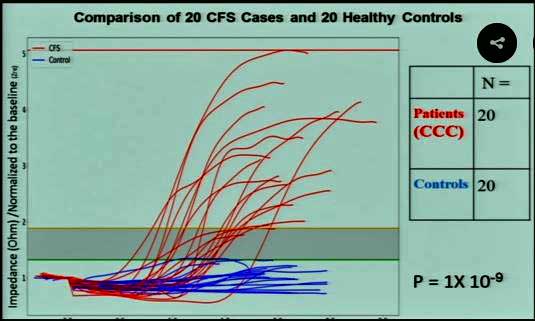
Nanoneedle
Meanwhile, on a brighter note, the nanoneedle continues to demonstrate an amazing specificity in ME/CFS. A quick look at the chart indicates a huge separation between the ME/CFS patients (in red) and the healthy controls (in blue). Statistically, the odds that such a thing could be happening by chance is now in the 1 in a billion range. We should see the results soon in the official journal of the National Academy of Sciences – “The Proceedings of the National Academy of Sciences of the United States of America”, a distinguished journal indeed.
Plus, the fascinating plasma issue is holding true. Adding ME/CFS plasma to healthy cells causes the impedance levels to wildly increase, while adding healthy plasma to ME/CFS cells causes them to react normally. Something in the blood appears to be taking a two-by-four to our cells. Studies indicate that it’s not a metabolite – our metabolism is not producing some metabolic breakdown component that is disturbing our cells. Nor does it appear to be a cytokine, but in what may be a very lucky break, Davis was able to recreate the problem with an exosome. Davis wouldn’t commit to it being an exosome, but right now that’s what it looks like. That would be a great break and good news, given that both Maureen Hanson and Prusty are examining exosomes.
We know the nanoneedle indicates something dramatically different happens to ME/CFS patients’ cells when they’re stressed with saline, but we still don’t know what – a question that clearly has to be answered. Davis suggested the increased impedance signal may reflect a mitochondrial breakdown, and that would surely make sense, given what we’ve learned. Two drugs – both coming from Bob Naviaux – that are able to obliterate that signal (Copaxone and an SS-peptide) point to that.
Then came a preliminary but really weird finding – elevated levels of mercury (not so weird) and high levels of uranium (really weird) associated with low selenium levels in the hair analyses of a significant subset of patients. Both the mercury and uranium issues derive from low selenium levels. Selenium, interestingly, also plays a role in the conversion of T4 to T3 in the thyroid. (Chris Kressler recommends getting your selenium levels tested and, in general, using dietary methods (not selenium supplementation) to safely increase selenium levels in hypothyroidism.)
A Common Theme?
A lot of the work was preliminary but it was fascinating to see the potential overlaps emerge: the intramural studies’ early mitochondrial findings, Lipkin’s possible plasmalogen and epigenetic iron findings, the mitochondrial breakdowns that Prusty induced using ME/CFS plasma or HHV-6A reactivation, and Ron Davis’s nanoneedle and plasma exchange results pointed to a general theme – mitochondrial issues and problems with oxygen delivery/utilization – which come to think of it, showed up on Day 1 with David Systrom’s, Maureen Hanson’s and Betsy Keller’s presentations. Plus, a new possible culprit has emerged – exosomes – which Maureen Hanson and Prusty are looking into now.
Check out the first day of the conference
Keep the Information Coming!
Support Health Rising


Thanks for this helpful synopsis, Cort. I found Dr. Levine’s presentation really interesting and was disappointed they didn’t give her more time. Indeed, I think the conference would have been stronger had *everyone* on that panel been given twice the time (at least) that they were given. Maybe the next time…
That was a really tough subject to cover in 10 minutes! You can find a larger overview of MCAS in this blog
https://www.healthrising.org/blog/2018/07/18/2018-dysautonomia-international-pots-sfn-mcas-vagus-nerve/
Thank you so much Cort for the summaries, it is so much appreciated!
Cort – Thank you once again for such an amazingly comprehensive yet succinct summary! Given that few ME/CFS sufferers have biochemistry PhD’s you provide an invaluable knowledge translation service to our community. We are fortunate to have you amongst us and I hope that as many people as possible will contribute whatever they can to keep you rolling down the road. We need you!
Thanks Joe. Contributions help 🙂
Hi Cort,
Thanks for another excellent write-up!
Possible confusion when you write “The fact that the reduced ratio of bacterioides/firmicutes bacteria found in ME/CFS…”
-> The label on the graph above it does seem to indicate that the ratio of firmicutes/bacteroides is reduced, but then again the text next to it does mention the bacteroides/firmicutes ratio is altered (without specifying up or down). -> Confusing slide.
I had trouble with that one. I had it one way and then changed it. I would go with the graph!
Dear Cort,
Thank you ever so much for all this hard work; you really deserve some huge recognition for this. Thank you.
I would like to add some observations, however, to one of the statements made by one of the physicians. The orthostatic hypotension problems are not really low hanging fruit, easy to pick (i.e. correct). If this were the case, then thousands upon thousands of patients would be a little more comfortable.
Firstly, Mestinon was used decades ago by Dr Holtorf and Dr Teitelbaum. You may recall they had these Fatigue and Fibromyalgia centres in about 4 or 5 cities across the usa. Mestinon use in this illness is not a new discovery; it is alas just reinventing the wheel.
Furthermore, patients are often very reactive to these types of drugs, as they produce (for some reason I do not understand) severe side effects. The typical cfs/me patient tries them all: midodrine, florinef, salt pills, fludrocortisone, vasopressin, effortil, (I may have forgotten a few others) and then has to abandon them because of side effects.
Another problem also arises: let us say a given patient does somehow tolerate one of these meds, and then this person starts to move about a bit, well, you can be sure they will at some point have a huge huge crash, or relapse. The reason, is that the fundamental problem in this illness has not been addressed. Correcting one symptom does not lead to a necessarily better life. In fact, patient will report that when they have been ‘spiked’ up on meds, they then later had huge relapses for a long time.
It might be useful for some of the doctors/researchers to also talk to some of the other doctors who treated this illness like Dr Teitelbaum, or Dr Holtorf. They have lots of experience, decades of experience. I am so happy that Dr Peterson is on the team.
I worry that sometimes something gets presented as new, and it is in fact, not new at all.
I so want a breakthrough, and the best minds, really best minds, need to be brought together form all over the world.
This is one of the worst illnesses on the planet.
Thank you every so much.
Maybe not low enough? I took Dr. Bateman’s words on that but my partner, if she doesn’t mind me saying. has POTS and, like you and others, reacts poorly – actually spectacularly poorly – to all POTS drugs.
Your experience reminds me of Dr. Cheney’s statement – I push patients towards health and their bodies push them back again. I’ve had instances where things helped at first and then sent me into a relapse…
Have you tried Ivabradine? Grubb at the last Dysautonomia conference, said it worked in 75% of POTS patients. He seemed very high on it.
Cort, your conversation with Perrier is hitting exactly the issues I am concerned/thinking about. Thanks you two for helping me to feel not so alone.
“Diet does have an effect on the microbiome, and it may play a role in the therapeutic approach to ME/CFS. (It’s hard to believe that it could play a significant role in triggering it.)”
I am not convinced it’s hard to believe that it could play a significant role in triggering it.
Last summer I learned that I have an extensive set of food intolerances. It took so long to discover this as I am intolerant to fructose (very strong) and to the whole FODMAP range (strong) plus some other stuff (less clear).
(Only) temporary avoiding one or two food categories did not make any noticeable difference as I am intolerant to about half of what I used to eat so I kept unknowingly eating plenty of food I was intolerant too each time.
Now this fructose intolerance seems to be caused by a (not diagnosed but it has very strong characteristics of it) fructose malabsorbtion. As I was unaware of it and fruit is supposed to be really healthy, I ate year round a lot of it from my childhood on.
With fructose malabsorbtion, plenty of it arrives at the large bowel. But fructose is a really “fast” sugar. It provides very “quick” fuel that can cause some species in the large gut to grow so fast they can outgrow all the others.
Having very fast multiplication rates and being able to find a food source some species grow a lot better on then other species should totally change the gut bacteria distribution.
One can say “well, fructose malabsorbtion is something specific and sort of a disease / dysfunction all by itself”.
I’m not sure. Fructose is structurally very similar to glucose. But glucose can be uptaken very easily in about any human. Fructose has a far reduced rate of uptake compared to glucose again in any human. In any human, fructose gets better uptaken as long as the ratio fructose/glucose is less then 1. Then there is something called a fructose/glucose shuttle where each molecule of glucose makes it easier to uptake a molecule of fructose.
So what is the difference between a healthy person and a person with fructose malabsorption? While fructose is a lot harder to uptake then glucose in both, it gets problematic in people with fructose malabsorption.
When one thinks about it, it’s an odd situation. There seems IMO to be very little chemical reason why fructose absorption is so much more complicated then glucose absorption. Yes, we do have better chemicals for glucose absorption. But my point is that I don’t see why we need so much more complex chemistry to uptake fructose then glucose.
That raises the question: is it on purpose (meant to be) that fructose uptake is so much more complicated then glucose uptake?
There are good reasons to be found for it:
* In nature, fructose/glucose ratios > 1 are near only found in ripe fruit.
* Crossing the fructose/glucose ratio of 1 is the tipping point of far more difficult uptake of fructose in (near?) all humans.
* During fall, ripe fruit is overly abundant.
* As fall comes before winter, humans in the past wished to eat plenty and plenty of an abundant food source rich in energy.
* If all that combined fructose and glucose load would have quickly digested in ancient times it would:
– create a very high blood sugar load. While it is often said only high glucose is bad, fructose makes blood just as viscous (thick, poor blood flow) as glucose IMO so that is pretty bad
– create a massive inflammatory reaction as the body would work full speed to decrease this sugar load in the blood by converting it to body fat. In ancient times producing body fat before winter would be good, but doing it by converting fructose to fat in the liver would cost huge amounts of NADPH. That’s needed for both creating fat from sugar and for recycling glutathione, the bodies main anti-oxidant. That in result would crash glutathione reserves and provoke high oxidative stress.
* Having a mechanism to “bypass / offload” this strong fall load of ripe fruit could be done by a mechanism that has a detection mechanism for ripe fruit. Like there being a fructose/glucose ratio higher then one.
* As some fruits dangle around that ratio and when one eats a lot of fruit then the combined fructose/glucose load can still be very high. Therefore setting a maximum uptake of fructose would be a good thing too.
* In a !healthy! gut, that big load of fructose reaching the large bowel could be converted by the gut bacteria to buteric acid (butyrate).
* Such a hypothetical mechanism would in fall decrease blood sugar spiking, decrease oxidative stress spiking (and inflammation too due to lesser need of insuline), would create plenty of anti-inflammatory butyrate that is good at countering the remaining inflammation due to “ripe fruit sugar overload” (even with this mechanism the blood sees still a lot more sugar inflow during fall) and plenty of butyrate would be present at the gut helping to repair it at times of increased body wide inflammation (inflammation and oxidative stress are believed to be “aggressive” to the gut, so reparation may be needed). While the fat yield would be a bit lower by letting the bacteria transform fructose to fat, it still should yield plenty of winter fat.
=> In short, poor fructose absorption in the small bowel is nowadays considered a dysfunction, but it seems to be at the same time an utmost outstanding mechanism for being able to convert large amounts of fall fruit into needed winter fat in a non-industrial society.
I consider it an evolved beneficial feature that helped humanity most of its history. So what’s different now? Diet! We only have since a few generations year round access to plenty of ripe fruit.
Before humans started at the beginning of fall with bacteria being fed “slow carbs” for months, likely producing much butyrate from any carbs. Feeding them so much ripe fruit will have changed their composition after many weeks. But winter and spring starved them out. Slow bacteria were bound to dominate again.
Now, we can develop specialized (fructose) gut bacteria that can survive and thrive all year round. No seasonal cleaning. They can evolve over many years to increase their grip on our guts. If our mothers where eating plenty of fruit year round these bacteria can be transferred from mother to child and further specialize into something the human gut has never seen before.
People with (strong or moderate compared to healthy people) fructose intolerance may be the canaries in the coal mine showing what’s to come with this changed and supposedly healthy diet. They are the first to develop this hypothetical strongly altered gut microbiome.
As to my FODMAP intolerance? Once these new fructose loving species settle in, they’ll eat moderate-speed carbs too. Slower then the specialized ones, but the numbers of the new fructose-loving bacteria are so great that they’ll still out-compete the more specialized ones when feeding them FODMAPs, “fibers” and sometimes even “slow fibers (resistant starch)”.
So in this idea does one needs to have fructose malabsorption to get gut dysbiosis or even ME? No. But someone with it loving to eat unknowingly fruit year round may not need much to get ME. Maybe such person may be more keen to be kicked into gradual onset ME when encountering small additional problems.
Someone with higher but still limited (for all humans it is limited) fructose absorption rates and or less habit to eat a lot of fruit may need a far stronger trigger like a strong viral infection in order to get ME.
Once ME started, blood flow gets disrupted. The gut is the first to get reduced blood given in times of disease and in times of reduced blood flow. That should reduces all absorption rates in the small gut, including fructose absorption rates. In this model, once ME is started people shift nearer to fructose malabsorption if they hadn’t or get deeper into it if they had before getting ill. This could well create part of a hard to break vicious circle always seen in ME.
Under this hypothesis, a supposedly healthy diet with plenty of fruit could trigger deep trouble for those predisposed or those getting an additional hit. Having the food industry adding cheap and sweet fructose to near everything including meat does not help matters.
As to people who lived for millennia on tropical places without winters? If what I learned from Geographic channel is wright sugary carbs are a rare thing even in much of the tropics, making raw honey a rare treat.
Hi Dejurgen,
I did a Metabalon test thrugh Dr. Kogelnik’s office, which showed my fructose levels are off the charts. The crazy thing is that I eat ZERO fruit. I do eat quite a few vegetables, like bell peppers. The PHD nutritionist out of Kogelnik’s office thought it was interesting, but she didn’t have any ideas about changing those levels other than not eating things with Fructose.
Curious if you have any insight?
Thanks for your time!!
Marc
Wow…We always seem to break the mold don’t we?
The first thing to look out for: did you get tested for hereditary fructose intolerance?
https://en.wikipedia.org/wiki/Hereditary_fructose_intolerance
“Hereditary fructose intolerance is an inborn error of fructose metabolism caused by a deficiency of the enzyme aldolase B.[1] Individuals affected with HFI are asymptomatic until they ingest fructose, sucrose, or sorbitol. If fructose is ingested, the enzymatic block at aldolase B causes an accumulation of fructose-1-phosphate which, over time, results in the death of liver cells.[1] This accumulation has downstream effects on gluconeogenesis and regeneration of adenosine triphosphate (ATP).[1] Symptoms of HFI include vomiting, convulsions, irritability, poor feeding as a baby, hypoglycemia, jaundice, hemorrhage, hepatomegaly, hyperuricemia and potentially kidney failure.[1] While HFI is not clinically a devastating condition, there are reported deaths in infants and children as a result of the metabolic consequences of HFI.”
“Because of the ease of therapy (dietary exclusion of fructose), HFI can be effectively managed if properly diagnosed. In HFI, the diagnosis of homozygotes is difficult, requiring a genomic DNA screening with allele specific probes or an enzyme assay from a liver biopsy. Once identified, parents of infants who carry mutant aldolase B alleles leading to HFI, or older individuals who have clinical histories compatible with HFI can be identified and counselled with regard to preventive therapy:”
“Older patients with HFI typically self-select a diet low in fructose, even before being diagnosed.”
-> You eat ZERO fruit you said? Being older…?
Problem with eating *only* zero fruit if HFI is really bad: dietary exclusion of foods containing fructose, sucrose, or sorbitol.
-> sucrose converts to half of it’s weight in fructose
-> sorbitol is often added in “light” products (sold sometimes as “zero sugar”)
-> fructose is added to near everything, going from meat over spaghetti sauce to mashed potatoes, especially if you live in the States
If not tested that would be my first and strongest guess.
“If you’ve ever disliked eating broccoli, artichokes, asparagus or okra, you’ll be delighted to know that they’re high-fructose foods”
“As you might guess, tomatoes and sweet red peppers are abound with fructose.”
“Other high-fructose vegetables are okra, mushrooms and peas.” (Parsnips, sweet potatoes, white potatoes, carrots and winter squash are hearty alternatives without the heavy fructose load.)
“Finally, instead of using high-fructose onions, leeks and shallots to add flavor to a dish, opt for low-fructose chives.”
-> probably sweet onions are worse then non-sweet onions but I’m not sure; better avoid them for a time if you have tested positive for HFI until blood values are under control.
“Avoid wheat — one of the major fructose foods — and any products made with it. This includes foods such as pasta, couscous or wheat bread. Products made with corn, oats, rice, rye or quinoa provide lower-fructose alternatives, as long as they aren’t sweetened with high fructose corn syrup. Tofu and legumes are low in fructose, but can also cause intestinal gas.”
-> I guess you do eat bread or maybe drink bear?
-> So there are far more products that contain fructose then just fruit. Source https://www.livestrong.com/article/263938-vegetables-fruits-that-contain-fructose/
Note: If not tested positive for HFI there are enzymes that can convert fructose to glucose back and forth but that is an unlikely cause IMO. If so, trying to avoid “combined” sugar blood spikes might help a bit.
Thanks Dejurgen!!! Really appreciate the response.
Im really pissed off that my doctor didn’t suggest testing specifically for HFI. Its kinda mind boggling to me.
Hey Cort! When you say “Wow…We always seem to break the mold don’t we?” Are you referring to this being rare or something you haven’t heard of for someone with ME?
We just often seem to have these odd test results which don’t seem to fit into the traditional categories. As a group I think we surprise a lot of people.
Good luck with the HFI test!
Hi Mark,
I am far from an expert but one thing that pops into my mind is that changing diet now might effect test results if it’s not a dedicated DNA test.
It may be important to ask that and decide based upon the answer (if diet would influence the test) if you want to correct the fructose values asap or if a correct diagnosis is more important.
Logic tells I should do such a Metabolon test too, but due to experience I went “test light”.
Having to beg for a test, getting “that look”, having to gather information myself on where and how to best do the test, paying for it and often ending up with not much more then a puzzled doctor even if tests show up weird values isn’t that a satisfying process :-(.
Good luck, whatever that may be!
“The blood and serum were a wash. The PBMC’s were a bit better, but when he looked, there wasn’t that much there there – even when the virus was present – the loads were low.”
“Prusty then looked for evidence that his mystery RNA was present in blood clot cells and found it – in about 40% of patients and in none of the controls.”
=> dr. Prusty may have a really BIG winner here!
I know someone who hasn’t got ME but shares quite some symptoms with it. Difference is, his allergen markers are sky high and his hart rate is once extremely high and another time ridiculously low.
As the first doctor that treated him was really concerned about his hart, he gave him a drug to stabilize his hart rate. To the doctors surprise his allergen markers rose even a lot higher. The best the doctor could come up with is that he maybe was allergic to the drug. I call it possible, but quite unlikely. As the patient says himself “I do find it strange, I am allergic to nothing else”.
I recently talked to him. He said he had from time to time huge leg spasms. I asked him a few question as I maybe had some similar experiences.
I asked him if his feet were shaking alternatingly at a very fast rate he could not do willingly. He said “yes, that’s it”.
I then asked him if his hart rate was very high at the moment of the spasms. He said no. Then he continued “it used to be before the drug fixing my hart rate. Then it rose to 200 bpm. When everything was over it then fell below 40 bpm. Now it remains stable.
I then asked if his legs were very cold at the time of the spasms. He didn’t know exactly, but he said his legs were as white as porcelain. They had absolutely no color since he started to get ill. That may be a clue to the answer being maybe yes.
He then said something about having several wounds. I asked him if it did bleed surprisingly few (since I have ME I barely lose any blood when cutting myself moderately deep). He said “yes, now you say it, there is surprisingly little blood going with even deep wounds”.
=> So what is my hypothesis?
When blood flow (returning of blood to the hart) gets very bad, hart preload is suffering a lot. As with any pump, increasing output pressure is done by “simply” pumping harder. For the hart, that’s contracting stronger.
But the inlet is a different thing altogether. When resistance in the inlet “pipes” is too high flow can only be increased by increasing the pressure difference. But the pressure at the muscles is limited to the pressure of air plus a little bit more from the skin acting like compression stockings. Combined that’s a bit above 1 bar (1 times atmospheric pressure). At the other side of the “pipe” the pressure can not go below zero. But even at pressure halfway the muscles pressure and zero there is a risk of creating a vacuum in the hart. At reduced pressure water starts to cook at body temperature, and so does blood. Blood cooking, even at body temperature, is a *very* bad thing.
So in order to avoid the risk of creating this vacuum (blood cooking) the hart has either to be controlled very precisely or either not expand a lot to be sure to be on the save side. But if it expands far less, fewer blood can flow into the hart and be pumped per hart beat. So in order to get a somewhat acceptable blood flow, hart rate has to go up *a lot* if the hart would chose this strategy.
Now I started to suspect that potentially this person has really poor (returning) blood flow in the legs. That *could* get his hart into small but very fast hart beats at times he got into trouble as he reported. Very fast hart beats are helped by dumping very large loads of adrenaline and *could* leave his adrenaline exhausted after the event, causing his hart rate to fall deep.
Now if that where to happen, and *IF* he had an allergic reaction to something clothing his veins and arteries (*very* high allergen values), then taking a drug that fixed his hart rate would prevent this surge in hart rate to make his blood return at somewhat acceptable rates from his legs to his hart. That would further reduces blood flow in the legs and leg temperature compared to not taking the hart drug. Both would increase blood clothing at the legs. Now if the allergic reaction was to the blood cloths itself… …the rise in allergen markers would be logical. I didn’t told him my thoughts as he is in the process of many clinical investigations and I don’t want him to take vital decisions based on a guts feeling.
But combined with dr Prusty’s “Prusty then looked for evidence that his mystery RNA was present in blood clot cells and found it – in about 40% of patients and in none of the controls.” it kinda feels like the good doctor struck true gold.
Blood cloths can be “dissolved” IMO by:
* immune cells “eating” the clots
* after some searching : “Mast Cells – release heparin, which prevents blood clotting and histamine, which promotes reactions (allergies and inflammation)” from https://www.coursehero.com/file/p6sef2e/Mast-Cells-release-heparin-which-prevents-blood-clotting-and-histamine-which/
Now mast cell activation (heparin) wouldn’t clean up blood cloths, but rather prevent / reduce blood clot forming. That would help too to have less overall blood clots.
Potential problem with this is, if the “size” of the problem is big enough, that the histamine promotes allergic reactions and inflammation. Inflammation, like potential immune cells “eating” blood cloths, produce often considerable amounts of oxidative stress.
Oxidative stress is good at killing NO. NO is needed to dilate blood vessels. Oxidative stress, if lasting a long time, can make RBC less flexible reducing blood flow in the capillaries a lot. Reduced blood flow coases lower body temperature and increased blood clotting.
If the inflammatory reaction is too strong, the body can be overwhelmed. Strong anti-inflammatory mechanisms have to be activated. I strongly tend to believe that one such mechanism is, when strong inflammation lasts longtime, to reduce blood volume. All “purple-ingrown-nail-toe” like inflammation has a lot of blood colored swelling. With few blood, that seems hard to achieve body wide so it seems to be very effective at reducing this particular type of chronic inflammation. Being long time inflamed over large parts of the body like a swollen purple balloon doesn’t seem healthy at all, so reducing blood volume to prevent this may be the better of two bad things.
Now reduced blood volume unfortunately isn’t going to help the supposed original problem: blood clothing. So now we have a potential strong vicious circle. With poor blood flow, low blood volume, high body wide immune activation, confusing both high pro- and anti-inflammatory markers, a strong sense of inflammation without visually seeing any inflammation, hart rate problems, strong fatigue and exercise intollerance, orthostatic intollerance, quick onset of anaerobic mechanism and lactate buildup, potential mast cell activation and increased allergen values or increased amounts of allergies…
Sounds like an extensive subset of ME to me. With few difficulty we can weave in turning down the mitochondria as a result of the very high oxidative stress the above generates. And breaking down of intestine integrity, the brain-blood-barrier, poor control of muscles and so on getting an even stronger vicious circle started.
If one truly wants to add EBV or other Herpes type virusses in this hypothesis, they are known to be able to increase baseline oxidative stress levels. This could lower the threshold for setting this mechanism into motion. As to very few specific immune markers: what type of immune marker does one expect against massive amounts of small (near microscopic) blood clots? They may not be the usual suspects. When the collateral damage gets too high then auto-immunity against for example blood vessels may show up, as seen in a large portion of patients according to recent research.
Something still felt missing: why is there so much “mystery RNA” in the blood cloths and not in the blood and serum?
The answer could be NETosis https://en.wikipedia.org/wiki/Neutrophil_extracellular_traps:
“NETs allow neutrophils to kill extracellular pathogens while minimizing damage to the host cells.”
“NETs disarm pathogens with antimicrobial proteins such as neutrophil elastase, cathepsin G and histones that have a high affinity for DNA.”
-> RNA ain’t DNA, but it does resemble it. And Herpes virusses (even latent) produce plenty of it and pump it in the bloodstream continously.
“More recently, it has also been shown that not only bacteria but also pathogenic fungi such as Candida albicans induce neutrophils to form NETs that capture and kill C. albicans hyphal as well as yeast-form cells.”
-> Maybe they can capture the highly toxic mold spores too?
“While it was originally proposed that NETs would be formed in tissues at a site of bacterial/yeast infection, NETs have also been shown to form within blood vessels during sepsis (specifically in the lung capillaries and liver sinusoids)”
-> Maybe they can capture the highly toxic mold spores, in the bloodstream, too?
-> As it has only recent been shown in specific tissue, it may be only there or more tissues may be added to this list in the future.
“NETs might also have a deleterious effect on the host, because the exposure of extracellular histone complexes could play a role during the development of autoimmune diseases like systemic lupus erythematosus.”
“NETs have also been reported in the colon mucosa of patients with the inflammatory bowel disease ulcerative colitis.”
“NETs are able to capture and kill various immune cell groups such as CD4+ and CD8+ T cells, B cells, and monocytes.”
-> Quite a far stretch, but could this help capture EBV infected immune cells, apart from the viral RNA they release?
Maybe also of importance:
“Azurophilic granule proteins such as myeloperoxidase (MPO) and…”
-> So they release myeloperoxidase, so I looked into https://en.wikipedia.org/wiki/Myeloperoxidase:
“MPO is a member of the XPO subfamily of peroxidases and produces hypochlorous acid (HOCl) from hydrogen peroxide (H2O2) and chloride anion (Cl−) (or the equivalent from a non-chlorine halide) during the neutrophil’s respiratory burst. It requires heme as a cofactor. Furthermore, it oxidizes tyrosine to tyrosyl radical using hydrogen peroxide as an oxidizing agent.”
Now hypochlorus acid is not only an oxidant, it’s a prime disinfectant (bleach). Disinfectant as in “kills things efficitently”.
See https://en.wikipedia.org/wiki/Hypochlorous_acid:
I’ll give the “short” version, it’s *really* interesting to read how hypochlorus acid could be involved in ME
“Hypochlorous acid reacts with a wide variety of biomolecules, including DNA, RNA,[8][15][16][17] fatty acid groups, cholesterol[18][19][20][21][22][23][24][25] and proteins.[21][26][27][28][29][30][31]”
“Knox et al.[29] first noted that HClO is a sulfhydryl inhibitor that, in sufficient quantity, could completely inactivate proteins containing sulfhydryl groups.”
“The polar chlorine disrupts lipid bilayers and could increase permeability.[19]”
“Escherichia coli exposed to hypochlorous acid lose viability in less than 0.1 seconds due to inactivation of many vital systems.[13][37][38][39][40]”
-> Just added as an indication on how poisonous the stuff can be.
This is a major one, an entire chapter called “Inhibition of glucose oxidation”
The exact mechanisms aren’t entirely clear, but research indicates it would both disrupt glycolysis and mithochondrial respiration as seen in ME:
“In 1948, Knox et al.[29] proposed the idea that inhibition of glucose oxidation is a major factor in the bacteriocidal nature of chlorine solutions. He proposed that the active agent or agents diffuse across the cytoplasmic membrane to inactivate key sulfhydryl-containing enzymes in the glycolytic pathway.”
“The question of loss of glucose oxidation has been further explored in terms of loss of respiration. Venkobachar et al.[46] found that succinic dehydrogenase was inhibited in vitro by HOCl, which led to the investigation of the possibility that disruption of electron transport could be the cause of bacterial inactivation. Albrich et al.[8] subsequently found that HOCl destroys cytochromes and iron-sulfur clusters and observed that oxygen uptake is abolished by HOCl and adenine nucleotides are lost. It was also observed that irreversible oxidation of cytochromes paralleled the loss of respiratory activity.”
“Later studies[38] revealed that Ubiquinol oxidase activity ceases first, and the still-active cytochromes reduce the remaining quinone. The cytochromes then pass the electrons to oxygen, which explains why the cytochromes cannot be reoxidized, as observed by Rosen et al.[40] However, this line of inquiry was ended when Albrich et al.[26] found that cellular inactivation precedes loss of respiration by using a flow mixing system that allowed evaluation of viability on much smaller time scales. This group found that cells capable of respiring could not divide after exposure to HOCl.”
There is also a topic on “Depletion of adenine nucleotides”:
“the observation that HOCl causes intracellular ATP hydrolysis. It was also confirmed that, at bacteriocidal levels of HOCl, cytosolic components are unaffected. So it was proposed that modification of some membrane-bound protein results in extensive ATP hydrolysis, and this, coupled with the cells inability to remove AMP from the cytosol, depresses metabolic function. One protein involved in loss of ability to regenerate ATP has been found to be ATP synthetase.[27] Much of this research on respiration reconfirms the observation that relevant bacteriocidal reactions take place at the cell membrane.[27][44][48]”
-> ATP-loss? Sound like ME too.
Another “nice” topic “Protein unfolding and aggregation”
“HOCl is known to cause post-translational modifications to proteins, the notable ones being cysteine and methionine oxidation. A recent examination of HOCl’s bactericidal role revealed it to be a potent inducer of protein aggregation.[51]”
-> unfolding and post-translational modifcations of proteins??? The bodies first defense is to break down the protein in the cell and redo the work. That costs additional energy and tones down metabolism (hibernation like). The second defense if that fails: NETosis!?!?! !producing more myeloperoxidase and hypochlorous acid doing more of the above, a vicious circle!
-> Protein aggregation? More NETosis and other immune cell activation. Yay!
“Hypochlorites are the salts of hypochlorous acid; commercially important hypochlorites are calcium hypochlorite and sodium hypochlorite.”
-> Both *might* be formed into the body and removed by urine. Is anyone testing for this stuff? Many mainly female patients see increase calcium loss compared to healthy controls. Near all patients have reduced blood volumes; that might be partially related to removal of sodium in the form of sodium hypochlorite. I have to add both ideas are very hypothetical.
“the notable ones being cysteine and methionine oxidation.”
-> So hydrochloruous acid “kills” cysteine and methione while myeloperoxidase “kills” thyrosine. Increased “wrong” protein destruction will increase amino acid wasting. NETosis itself will require plenty of amino acids as it uses mainly proteins to do its job. Sounds like it could help deplete amino acid levels as seen in ME.
Now need sleep urgently. The wired and tired part of me took over, it’ll hurt a lot tomorrow and thereafter…
If I didn’t explained my thoughts clear: looks like dr. Prusty may have found in these 40% of patients blood clots with mRNA originating from NETs set up in capillaries aimed at capturing Herpes viral DNA that infected cells produce even when the virus is dormant. That could both set of a severe blood flow dysfunction by clogging the capillaries and create a nasty immune activation as well as blocking oxygen uptake. All of these combined seem to have many potential vicious circle mechanisms.
A strong one shot infection or potentially mold spores (needs research for confirmation) may activate the same runaway mechanisms/disease.
Dejurgen, please see the following article about NETs, bicarbonate and CO2 levels (and thus indirectly O2 uptake) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5145884/
Lowering bicarbonate levels will hopefully allow the body to upregulate the metabolic processes that produce CO2. Once CO2 production is upregulated, O2 uptake hopefully will increase, allowing the body to balance redox reactions.
I’m currently trying to lower bicarbonate levels by supporting production of carbamoylphosphate from NH4+ and HCO3- by taking aminoacids (tyrosine, serine, glycine, methionine as SAMe, tryptophan as 5HTP) but am avoiding the aminoacids that are metabolised to alpha-glutarate or NO (glutamate, aspartate, arginine). I am hoping to upregulate the ureacycle and thereby lowering NO production through iNOS. Good results so far. Next I will try glutamine because glutamine seems to be the quickest source of NH4+ but I am hesitant as glutamine is metabolised to glutamate which in turn is metabolised to alpha-ketoglutarate. I might not be strong enough for glutamine yet.
Caveat: My body only accepted aminoacid intake after I got all B-vitamins, trace-minerals in place and electrolytes in place (especially NaCl). Before that I got ‘episodes’ which a doctor called ‘panic attacks’ but which could be attributed to an acid-base imbalance in the body, possibly respiratory acidosis corrected by metabolic alkalosis. This would lead to high bicarbonate levels unless the body again selfcorrects by downregulating CO2 production which in turn inhibits O2 uptake.
Hi Moira,
That certainly is an interesting link. I’ll have to reread it again and again to let sink in the implications however.
One difficulty is in assessing how to control bicarbonate levels independently from C02 levels in an in vivo buffer solution. The main property of a buffer solution (salt of the acid + acid in aquatic solution) is that it is able to keep the pH near constant in a wide range of mixtures (% of salt and % of acid).
In the case of bicarbonate/CO2 we have basically near all CO2 in the aquatic solution converting to either H2CO3 or NaHC03 assuming the most logical to use salt is a sodium based one. Very few C02 will be present in the solution.
In blood, that is different as much C02 will be bound to RBC, but this in vitro research does not mention their presence. CO2 bound to RBC will most likely behave totally different too.
So far, it remains unclear as to what they affect with “increasing the bicarbonate/CO2 ratio”. Do they mean initial applied bicarbonate versus surplus supplied CO2? If so, the additional CO2 should convert largely into bicarbonate itself in a real buffer solution as such is IMO the property of a buffer solution.
So is what they describe only a transient affect or an effect that can only exist locally and for some duration as long as conditions are ideal?
Another thing is that trying to adjust the strength of an immune response is quite a tricky thing to do. I must admit that I use to good effect Isoprinosine/Immunovar that is known to down-regulate (part of?) the immune system but I try to minimize its use and stay prudent on doing so.
But when I said that NETosis probably creates damage by itself that needs to taken care of by additional NETosis that does not need to mean a runaway thing. It can also be an amplified response that still is a “correct sized” response. A bit like breaking out a floor to place a new one in a house will create additional work like having to re-plaster the walls and grinding up the doors as the breaking did cause some extra damage. That doesn’t mean we have to leave one row of tiles next to the walls and doors in their original state.
Their are a few things that concern me regarding trying to raise blood CO2 levels:
* My body does a lot of effort to “hyperventilate” with one of the main results of doing so low CO2 levels. Is that collateral damage or the purpose itself?
* “We observed that both a low pH and a high CO2 to bicarbonate ratio decrease the capacity of neutrophils to release NETs.” -> If I understand correct the most reasonable thing to reach that (in steady state) in a buffer solution in vivo is to remove NaHCO3 (by disposal through urine) from the blood, not increasing CO2. Less Na be it NaCl or NaHCO3 or another sodium salt by disposal through urine should lead to less blood volume such as is seen in ME. (Still my NaHCO3 blood values seem to be just on the high half of the “good” range in my blood somehow; puzzling)
* “Live cell imaging further revealed that excessive bicarbonate induces chromatin externalization from neutrophils” -> Now again getting excessive levels of (Na) bicarbonate in a buffer solution would be the logical result of adding CO2 after some waiting time for the buffer to keep the pH constant by converting the additional H2CO3 that increased acidity and “move” it towards NaHCO3 IMO.
* Figure 2 shows a strong (temporary) increase in intracellular calcium levels under condition of increased CO2. With the increased acidity going with increased CO2 that makes sense again. IIRC however increased intracellular calcium levels go hand in hand with increased inflammation and can induce massive apoptosis/cell death. Not what I desire if correct.
* “LPS-stimulated NET formation was reduced under conditions of relative *hyper*capnia” goes somewhat against “Similarly, NET formation was also not pronounced at relative *hypo*capnia” so the effect of steady state CO2 levels of NETosis remains unclear to me.
*”The stimulatory effects of conventional agonists were markedly increased by a low pH and the neutrophils responded with more production of H2O2 and increased release of myeloperoxidase (13). A more recent publication confirms these results, indicating that a pH of 6.0 prolongs neutrophil survival and increases phagocytosis of bacteria” -> Having less NETosis (reducing capillary blocking) but more immune action from neutrophils (having more damage per immune action then NETosis) is kind of a tricky trade off IMO too. I am wary of that.
I’d love to see you getting this work, and get it work reliable and reproducible for all of us but I don’t understand yet how to get it done. I see several reasons to be prudent and watch out for both short and long term effects and side effects. As you seem to be very smart and methodological that will be no problem ;-).
Personally I am more drawn too
“In addition, we tested the influence of the bicarbonate to CO2 ration on the NET-inducing potential of monosodium urate crystals (MSU).” and the statement that monosodium urate induces NETosis.
-> That makes sense to me: uric acid crystals and crystals of their salt are known to be highly inflammatory and likely able to block capillaries. As uric acid is an anti-oxidant and NETosis creates very strong local oxidative stress, NETosis is a good way to break these crystals down. As I am in a line of men with increased uric acid production that is of interest to me. My values are below max, but they should be really low given how high I believe oxidative stress is in ME. I tend to believe in ME, with already poor blood flow, low enough uric acid values are even more important.
So for now I’ll stick to try and avoid the “need” for NETosis to kick into action. That includes:
* Avoiding infection (skin, airways, ears) decreasing both need for immune and NETosis activation
* Do small sets of circulation exercises spread over the day. Meant to counter circulation decreasing effects of NETs plus “grind down” blood clots and micro-scabs at arteries. Smaller debris should hamper blood flow less and require less immune action to break down (larger surface area versus mass for the immune chemicals to take effect needs less of these chemicals)
* Learn to breath well, to counter effects of poor oxygenation plus in the paper you provided it mentions increased NETosis through ischemia/re-oxygenation cycles. Also immune activity strongly increases when having these.
* Try and restore the gut. Major one for me. Means less partially digested food/proteins, toxins and bacteria enter the blood stream. Means less need for NETosis.
* Improve protein digestion (papaya, digestive enzymes). Means less need for NETosis when having leaky gut.
* Don’t overeat. Eating is an inflammatory process.
* Try and eat the correct food mix to fight oxidative stress.
* Pace, Pace, Pace… Means less blood flow disregulation and inflammation. Should reduce overall immune response and NETosis.
* Shift some protein sources from animal to vegetarian as I believe my uric acid levels should go down (in my case!).
* Salt is a big unknown to me. Undecided to either increase or decrease as implications go both ways.
Nothing new in the above, so I try to improve efficiency in doing so. Would be very happy if you can find a better approach ;-).
BTW: what sparked your initial interest to “aim” at NETosis? Could be interesting to learn.
Hi Dejurgen, the whole point would be to help the body get rid of CO2.
Red Blood cells:
The red blood cells have a double role
—a) carrying CO2 via hemoglobin to be excreted in the lungs and
—b) making HCO3- out of CO2 via the enzyme carbon-anhydrase (look up chloride-shift!). HCO3 – is then
—b1) excreted by the kidneys directly or
—b2) first metabolised to urea by the liver and then excreted.
So there are 3 ways that the body can excrete CO2: via the lungs, the kidneys and the liver.
Lungs:
In ME/CFS there seem to be high levels of free radicals which could lead of formation of methemoglobin which would give the blood a brownish appearance. Methemoglobin cannot transport CO2 to the lungs. That leaves carbon anhydrase to make HCO3- from CO2 to be excreted by the kidneys (directly) or by the liver (via urea).
Kidneys and Liver:
The kidneys and liver are required to work in parallel to keep the buffering system working properly. If for some reason one system can’t properly excrete HCO3-work, then the other system would also hold on to HCO3- in an effort to maintain proper pH levels.
NETs:
Eventually HCO3- levels would rise in the body and according to the article would induce (and maintain) NET formation.
Could continued NET formation long after the pathogen has gone explain the nanoneedle results of Ron Davis? I have no clue but find it interesting that Maureen Hanson is looking into exosomes.
To answer your question: A while ago I was looking into the results of the nanoneedle experiments of Ron Davis and looked up blood viscocity. I then looked up more info about exosomes as Maureen Hanson was looking into them. Combining blood viscocity and exosomes led me to find info about NETs so I was intrigued when you mentioned NETosis.
AminoAcids:
Metabolism of AminoAcids would support the liver in making urea (as a way to excrete HCO3- ) and thereby resolve the traffic jam of CO2 caused by methemoglobin which in turn is caused by oxidative stress.
Resolving the CO2 traffic jam would lower HCO3- levels in the body which in turn would stop the continued (unnecessary?) NET formationAll hypothetical. Will it actually work? Possibly not for everyone.
In my case I think the liver is the issue in CO2 homeostasis but in others it might be the kidneys (urate crystals?) and in some others somethings totally different. The point is that the liver, lungs and kidneys should be supported simultaneously otherwise when trying to help the body get rid of CO2 otherwise (to paraphrase one of Cort’s comments) the harder you push your body towards health, the harder it pushes back.
Your hesitation to take aminoacids is understandable as one interferes with the body’s buffering system which could be dangerous. However, hopefully the above explains better what I’m trying to do based on the latest research observations in ME/CFS and hopefully it will help someone else as well.
Hi Moira,
I totally misunderstood your previous comment. I thought you were to go against what the body seems to do and was going to try to increase blood CO2 values.
Better getting rid of CO2, I can feel the sense in that. Doing so by a process that is increasing ammonia in the body is a tat tricky as ammonia can cause brain swelling and we already have got high CSF pressure. I read you hope it to convert to urea in the liver, but before it gets there… who knows?
If I am correct, PaCO2 in ME patients is below that of healthy patients. IMO that already points to lower bicarbonate values, at the point of measurement being the main veins, compared to healthy persons.
Maybe the smallest capillaries that are under heavy stress might see very little flow and hence get very high CO2 values. That could cause that local and or transient behavior I spoke of.
It still is hard to determine for me what reducing overall CO2 values even further would do with the amount of NETosis in these places.
It’s still unclear to me if turning down NETosis would be a good idea as there might be a legit reason to keep it on. If so, not having it on may force a more damaging mechanism into action.
Trying to reduce it seems to me to bet on it that no other problems maintaining ME are left. In my case that is not so. In many cases I tend to believe that is a long stretch.
I more and more believe that ME is both a devastating disease and a mechanism not only keeping us alive in unlikely circumstances but even often doing so with remarkable few obvious permanent damage. We probably have worse blood flow then most diabetics in many places in the body, yet near all we keep our limbs and eyes without medication. Some unlikely combination of things seems to happen at these locations.
If I let talk my gut feeling, NETs in these capillaries may clean up blot clots with far less damage then any alternative mechanism can do. And it may be an essential and highest priority task. Blood sedimentation rates even near the max value would be an indication against trying to decrease NETosis in that view. But it is for sure nothing but a gut feeling.
Good luck and keep safe!
Hi Cort,
thank you for your excplenation! But what can I do laying here in bed in Belgium. If I am right, the only thing is the testing for OI. But then I have to be transported laying to the netherlands. And there they do not the 5 minutes NASA lean test but a 40 minute test! And they will never use the medications dr Bateman and Syström are using. So what can I do?
Many thanks for this great summary!
Wurzburg Würzburg
I’d like more information about the Precision Medicine Test for Pathogens.
I’m not sure which one he uses but this one popped up with I looked – https://www.healio.com/infectious-disease/practice-management/news/print/infectious-disease-news/%7bcd78e095-738b-4632-a693-3700f9c8ff2f%7d/genomics-soon-to-be-standard-for-rapid-id-diagnostics
how is it possible that nobody looks at the eds connection? it is not just a comorbidity, it is not jus worsening the me symptoms. 6 of 10 people with severe me do have some form of it.
It’s a biggie, I agree. ME/CFS experts are definitely aware of it but it hasn’t made its way into ME/CFS research so much. We certainly need to be aware of research developments in EDS…
Cort, you are an exceptionally gifted and passionate writer. Thank you for sharing this gift.
Great piece Cort. I am not surprised about the barren findings on viruses, it’s long past the point where we should have stopped wasting our time on that dead end.
The mitichondria sounds like a key area for focus.
A new disease outbreak is due to our mitochondria? Really?
Cfs is triggered by viruses. A virus or viruses do not perpetuate the illness.
We have had 25 years of research into viruses and cfs. I am very very confident that if a virus perpetuated cfs we would have found it.
Funds are limited. We need to focus on things that matter rather than going down the barren path of seeking ‘the elusive causative virus’.
No but as Prusty suggested an infection could possibly disable the mitochondria…(!)
Great write-up. Must admit I was sharing the lack of enthusiasm after the first portion of the dat 1 write-up. Not any more. Lots of good things happening and although it’s a slow creep, it definitely seems as though the right type of progress is being made.
Hi Cort,
Always interested in reading your uptodate information.
Sorry on another matter….
My daughter is soon to have a baby and asked me to get a Whooping Cough vaccination, of course I am worried that it may cause me to have a relapse of ME/CFS, as I am currently functioning 75% at the moment.
Any data or information I should read before.
I’m not intimately familiar with CFS research so it’s likely I’m missing something, but I often wonder if researcher are thinking about the lesser known type of mitochondrial dysfunction that causes excessive free radical overproduction while ATP output remains relatively unchanged. Generally when we think of mitochondrial dysfunction we think of a breakdown in the respiratory chain leading to elevations in certain metabolic byproducts and lowered ATP output, but mitochondria do more than produce energy, they also produce heat. Adaptive mutations make mitochondria LESS efficient allowing certain races of humans to survive better in colder climates. These mutations are a double edged sword though because the introduce genetic instability, which can result in mutations that lead to overproduction of reactive oxygen species and cellular toxicity.
There is an excellent talk by Dr Doug Wallace in the MitoAction.org podcasts where he outlines this process. The talk is an hour and forty minutes but starting at 1:16 he demonstrates how relatively minor genetic changes can result in mitochondria that show no significant reduction in ATP output and only a 20% reduction in oxygen consumption in a normal state, but under metabolic stress, oxygen consumption plummets, and instead of ATP being reduced like you would see in a classic mitochondrial disease, reactive oxygen species are massively overproduced.
Whether or not the cause is genetic, when viewed through the lens of this less known form of mitochondrial dysfunction, results like the Stanford study are not as confusing as they might seem. It also gives us a plausible hypothesis for the widespread HPA axis dysfunction and small fiber neuropathy that underlies the orthostatic effects of CFS. Due to evolutionary sympathetic bias, the parasympathetic nervous system is much more metabolically fragile and more vulnerable to oxidative stress. Whether the problem is the massive overproduction of ROS, or problems cleaning up ROS like you see in certain complex molecule diseases, as soon as ROS overwhelm the bodies ability to clean them out, oxidative stress takes place, leading to a vicious cycle of parasympathetic damage, which then increases sympathetic drive, which increases metabolism, which further increases ROS. Again, I’m not a scientist, but it seems like this mechanism could explain Jared Youngers findings of cerebral hypoxia leading to inflammation without involving the immune system at all.
Another mechanism that drives this sort of metabolic feedback loop is hypocapnia, (too little carbon dioxide in the blood). Hypocapnia can be a downstream effect of orthostatic issues or an effect of mitochondrial dysfunction directly and of course there are other causes. CFS patients have been found to be hypocapnic. The way I understand it, hypocapnia upregulates calcium flux in the heart, which then drives the heart harder than it should which places increased demand for ATP, thus depleting ATP faster, and when the heart can’t keep up with it’s energy demand on a cellular level the relaxation phase of the heartbeat is affected first, (which interestingly requires more ATP than contracting) leading to increased pressure in the pulmonary loop, which changes the ability of the lungs to exchange waste gasses because oxygen binds too tightly at increased pulmonary pressures, which then leads to more hypocapnia, more calcium channel up regulation, and more ATP depletion. It’s a vicious circle that is now thought to be a driver of heart failure.
I just wonder if researchers are considering these mechanisms in addition to some of the more exotic immune hypothesis. I’m sure better minds than mine have already considered this, I just don’t see it being considered so it makes me wonder.
Minor typo – you have “Kressler” instead of Kresser, at the end there.
But a really great write up, bringing all this information together. I’ve not been following developments for a couple years, so it’s interesting to see the research having advanced a little, overlapping and producing repeatable diagnostic results and bits and pieces that tally with my own personal testing. Exosomes sound interesting!