


Geoff’s Narrations
The GIST
The Blog
The GIST
- The second of a series of blogs on recent findings on the mitochondria in chronic fatigue syndrome (ME/CFS), long COVID, and fibromyalgia focuses on ME/CFS.
- The first ME/CFS mitochondrial study on the docket is “only” a case report – but what a case report! The senior author of the paper was none other than Alan Light, and that deserves a little digression.
- Light attempted to do for fatigue what had been done for pain; that is, delineate the fatigue pathways produced during exercise. Alan and Kathleen Light pioneered a series of studies in the 2010s which showed that ME/CFS patients’ white blood cells were hyper-reacting to metabolites released in the blood during exercise.
- The Lights went on to show that the response to exercise in ME/CFS and another fatiguing disorder was similar and different: both diseases demonstrated a dysregulation of the fight or flight or sympathetic nervous system, but only the ME/CFS patients’ white blood cells went into a kind of frenzy as they searched for signs that the muscles had been damaged.
- The Lights also participated in a pioneering heritability study in 2011 which concluded: a) there was a strong heritable contribution to ME/CFS (i.e. ME/CFS can run in families), b) a group of “high-risk” family lines were present (and should be studied). But the NIH, rather bizarrely, refused to provide further funding for it.
- After about 10 years of steady work on ME/CFS, the Lights last ME/CFS publication was in 2017. Then in 2024, Alan Light popped up in an intriguing case study involving a 75-year-old woman who’d been sick for over 20 years following an Epstein-Barr infection.
- A deep dive into her mitochondrial DNA and her mitochondria, however, revealed that she had several mitochondrial DNA mutations. The authors believed these mutations would be devastating and have “progressive effects on ATP production and mitochondrial functions”.
- They believed the herpesvirus infection triggered the mutations by producing large numbers of reactive oxygen species (free radicals) that already weakened mitochondria were unable to cope with.
- While we don’t ordinarily think of mitochondria as part of the immune defense, they play an important role in mounting the alarm when pathogens show up. In response, viruses produce large numbers of “virologs” in an attempt to disable the mitochondria and turn down the immune response.
- Next, a recent animal model explored how fatigue produced by the brain or “central fatigue” affects the mitochondria. The idea that central fatigue could be a big deal makes sense since ME/CFS symptoms are similar to the flu-like symptoms produced by the brain when we catch a cold.
- The study, which stressed mice but did not make them exercise harder, found that the stress damaged the mitochondria both in the brain and the muscles in several ways. That suggested that changes in the brain could affect the mitochondria in the muscles (!).
- The authors proposed that the fatigue, reduced endurance, and cognitive problems found in the mice with central fatigue were related to mitochondrial damage, problems with energy metabolism, and oxidative stress. Once again, they believed that mitochondrial damage played a key role in producing these symptoms.
- Finally, a very small but perhaps telling Stanford study found that stimulating the T-cells of ME/CFS patients resulted in increased numbers of dead and dying T-cells they believed might have been the result of damaged mitochondria which couldn’t “handle the heat” when they were tasked with getting activated.
- Once again we were back to the mitochondria. Whether in the brain or muscles or immune cells, these studies – all of them small, it should be noted – brought us back to what might very well be the original sin in these diseases: dysfunctional mitochondria.
 Health Rising’s Quickie Summer Donation Drive is On!
Health Rising’s Quickie Summer Donation Drive is On!The first ME/CFS mitochondrial study on the docket is “only” a case report – but what a case report! The senior author of the paper was none other than Alan Light, and that deserves a little digression.

Alan Light is back! He and his wife did pioneering work on ME/CFS in the 2010s.
Alan Light Returns!
We can’t just step over the fact that Alan Light has published again on ME/CFS. For those who don’t recognize them, he and his wife, Kathleen Light, are University of Utah pain researchers who, from 2009 to about 2017, produced a series of promising studies on ME/CFS.
Their 2012 paper produced perhaps the most startling graph I’ve seen in ME/CFS yet. It indicated that exercise produced remarkable increases in the expression of the receptors on white blood cells that detect metabolites associated with exercise. In essence, it suggested that exercise was either inducing a lot of damage which the white blood cells were reacting to, and/or that the white blood cells had become hypersensitive to any signs of damage produced by exercise. (Exercise always induces some damage). In either case, it appeared that an exercise-induced immune activation was in full bloom.
Interestingly, given Naviaux’s emphasis on the purinergic metabolites, the paper particularly plucked out the activation of a purinergic receptor (purinergic type 2X4 receptor). The paper also showed that when it came to exercise, people with ME/CFS and another notoriously fatiguing illness, multiple sclerosis (MS), were similar and different.
Exercise did not provoke the white blood cells in MS to go on high alert for exercise-induced metabolites, but it did trigger a spike in adrenergic receptor activity in both ME/CFS and MS patients (but not healthy controls). That suggested exercise dysregulated the sympathetic nervous system in both diseases.
Elucidating the Muscle Fatigue Pathways
The Lights’ study showed a dramatic increase in white blood cell reactivity to metabolites released during the muscles during exercise.
Alan Light said he wanted to do for fatigue when had been done for pain (i.e., elucidate the fatigue pathways in the body) and in 2015, he and Markus Amann put their findings together in a review paper, “From Petri dish to human: new insights into the mechanisms mediating muscle pain and fatigue, with implications for health and disease“.
The paper asserted that metabolites produced by muscle activity activated both myelinated and unmyelinated neurons (small nerve fibers) which then sent signals to the central nervous system via the nerve located on the dorsal horn of the spinal cord.
They noted that two sets of receptors designed to interact with these exercise-induced metabolites were present: one subtype responded to relatively low levels of intramuscular metabolites (lactate, ATP and protons), as seen during `normal’ (i.e. freely perfused and aerobic) exercise, while the other responded to higher concentrations of metabolites that were produced during ischemic conditions in which the muscle was in a state of hypoxia; i.e. low oxygen levels.
Over time, studies concluded that protons, ATP, and lactate produced by the muscles induce muscle fatigue and pain, and that neurons with ASIC, P2X, and TRPV1 receptors respond to them. The Lights found that exercise highly upregulated these receptors in ME/CFS. With that, they seemed to be on the road to delineating how muscle activity during exercise could be producing the pain and fatigue in ME/CFS, but the Light’s time with ME/CFS ended in 2017 with Dane Cook’s paper showing that exercise impacted brain functioning.
Inheriting Dysfunction (at the NIH)? The Heritability Study
The NIH funded an exploratory heritability study – and then when it proved to be successful – refused to fund a follow-up…
The Lights also participated in a 2011 paper, “Evidence for a heritable predisposition to Chronic Fatigue Syndrome“, that was remarkable for: a) its conclusion that there was “strong support for a heritable contribution” to ME/CFS (i.e. ME/CFS can run in families), b) its ability to identify a group of “high-risk” family lines, and c) the bizarre fact that it was never followed up on (they tried).
Identifying high-risk family lines was a coup, as researchers typically study families to get clues about what’s going on in a disease. The bizarre part comes from the NIH funding what turned out to be a successful study – and then being unwilling to fund a follow-up. (Why fund the study in the first place?)
What happened to turn off the Lights’ ME/CFS spigot is unclear, but the 2017 paper was the last one from the Lights and ME/CFS until 2024, when Alan Light popped up in an interesting case report.
A Viral – Mitochondria Connection?
The authors believe that an EBV infection probably produced the mutations found in her mitochondrial DNA.
The case report, “The “Mitochondrial DNA Missense Mutations ChrMT: 8981A > G and ChrMT: 6268C > T Identified in a Caucasian Female with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) Triggered by the Epstein–Barr Virus“, involves a 75-year-old woman who had been sick with ME/CFS, triggered by an infectious mononucleosis infection about 20 years earlier.
Her symptoms were primarily mainly neurological and cognitive impairment, and included fatigue, severe PEM, severe dysautonomia, unrefreshing sleep, widespread achiness and tenderness, sporadic dizziness with vertigo, severe orthostatic intolerance, and brain fog.
Standard lab tests, of course, were normal. The NASA lean test, though, revealed that she had neurally mediated hypotension (reduced blood pressure upon standing). Despite taking antivirals (valacyclovir, valganciclovir) for long periods, she at times exhibited moderately high titers of EBV antibodies. Still, neither, her antibody nor a PCR test suggested she had an active infection.
But then there were the mitochondria…
The electron chain complexes where ATP is finally produced are mainly encoded by mitochondrial chromosome DNA (ChrMT DNA, or mtDNA) passed down through the mother. The ChrMT chromosome is also where the first half of the mitochondrial energy production process – the Krebs cycle – where oxidative phosphorylation (OXPHOS) takes place. Because ATP generation produces a lot of oxidative stress (i.e., many free radicals) – picture sitting next to a blast furnace – this part of our DNA is highly susceptible to oxidative damage and has high mutation rates.
A deep dive into her mitochondrial DNA and her mitochondria, however, revealed that she had several mitochondrial DNA mutations (ATP6 (ChrMT: 8981A > G Q152R) and Cox1 (ChrMT: 6268C > T A122V)) including one at a site (ATP6) well known to cause mitochondrial problems.
That ChrMT Cox1 mutation was not just any mutation. For one, it was new to the literature. For two, it occurred in a gene (Cox1) that plays an important role in the later stages of the electron transport chain. The authors believed that a mutation affecting those stages would be particularly devastating and have “progressive effects on ATP production and mitochondrial functions”.
One of those effects was large numbers of dysfunctional extracellular mitochondria. These mitochondria appeared to “protrude” or “secrete” vesicle-like structures similar to those that Gram-negative bacteria produce in stressful conditions (!).
The authors proposed that the extracellular, abnormal mitochondria found in her and other ME/CFS and post-infectious illness patients probably resulted when a batch of malfunctioning mitochondria met up with a virus.

Sitting next to the blast furnace that is the mitochondria, it’s no wonder that mtDNA are more susceptible to mutations.
They also proposed that viral-induced reactive oxygen species (free radicals) were at least partly to blame for the mutations in the mitochondrial DNA found.
We’ve seen both of these hypotheses show up. They suggest that dysfunctional mitochondria not only fail to rise to the challenge when a pathogen shows up but break down and start producing large amounts of reactive oxygen species, which then produce more mitochondrial damage.
The authors proposed that people with ME/CFS probably had “dysfunctional mitochondria” that predisposed them to develop ME/CFS after viral infections”. Those dysfunctional mitochondria could either be inherited, or modified by epigenetic changes, and/or by an infection.
With regard to this patient, they believe her mitochondrial mutations probably reflected “accumulated mitochondrial stress” caused at least in part by her exposure to the Epstein-Barr virus at age 52.
Harkening back to the heritability study, they recommended, though, that her family undergo ChrMT DNA sequencing to “help uncover the role that viruses, mitochondrial DNA mutations, and mitochondrial problems play in the disabling fatigue found in post-infectious diseases”.
While we don’t normally associate the mitochondria with viruses, as Dr. Naviaux has explained, and as we saw in a recent blog, the mitochondria play a pivotal role in our viral defenses. Because they do so, they are targeted by viruses which produce large numbers of “virologs” in an attempt to damage the mitochondria and turn down the immune response.
The Brain-Mitochondria Connection
Let’s forget infection for a moment though. Could stressful conditions all by themselves lead to mitochondrial breakdown? A recent animal study suggests they can.
Several researchers have proposed that much of the fatigue in ME/CFS comes from “central fatigue”; i.e. fatigue that’s caused by the brain. In fact, Behan and Chaudhuri’s 2004 tome, “Fatigue in neurological disorders“, which repeatedly cited ME/CFS, was one of the first to make this connection. Their focus on the pathways interconnecting the basal ganglia, thalamus, limbic system, and higher cortical centres still rings true today.
The idea that central fatigue is a big deal in ME/CFS makes a lot of sense in several ways. For one, we know that the symptoms of “sickness behavior” we experience when we have a cold are produced by the brain. Given its typical post-infectious onset, ME/CFS could simply be a cause of chronic, unrelenting “sickness behavior”.
Of course, the big question is how to account for the findings of muscle damage, blood vessel, and mitochondrial problems that have shown up in the periphery in ME/CFS. Does the brain have that far of a reach?
A fascinating study from Daniel Clauw suggested it just might. In another animal study, Clauw found that increasing the activity of the insula – an organ of the brain involved in autonomic nervous system regulation and sensory processing – resulted in the development of small fiber neuropathy and increased pain sensitivity in its limbs (!).
The authors proposed that the fatigue, pain, sleep and cognitive problems caused by putting the mice under stress were due primarily to mitochondrial problems.
A recent animal model, “Replicating human characteristics: A promising animal model of central fatigue“, set out to understand just how far “central fatigue” might go. It first created a state of central, or brain-induced, fatigue via sleep deprivation and alternate-day fasting. Note that no increases in physical exertion were introduced – whatever happened in the periphery was not the result of exertion.
Even though the mice didn’t exert themselves, their muscles were hit hard. They exhibited reduced grip strength, and endurance, increased lactate levels and energy consumption, and muscle atrophy, including a reduction in slow muscle fiber levels.
And then there were the mitochondria. The mitochondria both in their brain and the muscles got hit even harder. Noting that mitochondrial problems have been found in another fatiguing disease – ME/CFS – the authors reported finding reduced ATP levels, increased levels of oxidative stress, irregularly shaped cells, increased heterochromatin in the nucleus, decreased levels of mitochondria in the cytoplasm, broken or disappeared inner cristae, and ruptured outer membranes.
In the end, the authors proposed that they believed that the fatigue, reduced endurance, and cognitive problems found in the mice with central fatigue were related to mitochondrial damage, problems with energy metabolism, and oxidative stress.
We were back to square one with the mitochondria – and a brain that apparently has a long reach.
Microstructural Mitochondrial Abnormalities
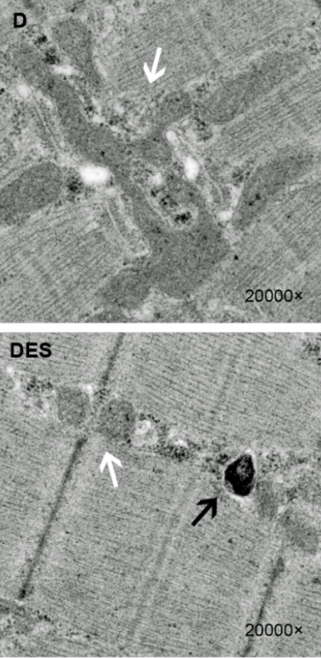
Swollen (damaged) mitochondria (white arrows) were greatly increased in the ME/CFS patients. (Image fr. Jingjing-Fan-Xiaoqi-Yang-Jie-Li-Ziyang-Shu-Jun-Dai-Xingran-Liu-Biao-Li-Shaohui-Jia-Xianjuan-Kou-Yi-Yang-and-Ning-Chen-CC-BY-3.0-Creative-Commons-via-Wikiimedia Commons)
A small but perhaps telling Stanford study from Fereshtah Jahanbani (“Phenotypic characteristics of peripheral immune cells of Myalgic encephalomyelitis/chronic fatigue syndrome via transmission electron microscopy: A pilot study“) used transmission electron microscopy (TEM) to dig deep in the microstructures (including the mitochondria) of both unstimulated and stimulated immune cells.
Only four people participated in the study – a pair of identical twins, one of which had moderate ME/CFS, a person with severe ME/CFS, and another healthy control – but the study produced some intriguing results that will hopefully be followed up on.
Immune activation is not a subtle process. It requires a lot of energy, and thus provides a nice way to test a cell’s energy metabolism. Placid-looking monocytes, for instance, turn into hairy monsters called macrophages, and B and T-cells jack up their energy metabolism as they prepare to clone themselves in great numbers.
Researchers have proposed that all that activity is too much for the puny mitochondria found in people with ME/CFS, causing the immune cells to punk out and either become exhausted (T-cells) or fail to reach maturity (B-cells).
The authors of this small study might agree. Once stimulated, the number of dead and dying T-cells in the ME/CFS patients began to pile up, and the levels of swollen mitochondria – indicative of mitochondrial dysfunction – increased significantly. The authors reported that the two ME/CFS patients “showed remarkably higher numbers of swollen mitochondria.”
The Original Sin – Bad Mitochondria?
Once again we were back to the mitochondria. Whether in the brain or muscles or immune cells, these studies – all of them small, it should be noted – brought us back to what might very well be the original sin in these diseases: dysfunctional mitochondria.
- Coming up: Pt. 3 – the Mitochondria in Fibromyalgia Plus Some Treatment Suggestions
Please Support Health Rising and Keep the Mitochondrial Information Flowing


Looks like we are really getting closer and closer to a nice story to explain ME/CFS and his main symptoms: PEM and brain fog.
Some people could be genetically predisposed to mitochondria mutations that could be triggered by stress (stressful life events) and/or virus (EBV here but maybe also the highly suspected HHV6-A).
Once ME/CFS is declared, the more stressed you are in your life and the more severe the symptoms you are going to have. This is a vicious circle.
All this would at the same time reconcile the different hypotheses and approaches to fix the problem. A medical solution (genetic, anti-viral, etc) combined to a ‘mental ‘solution like anything allowing to lower the stress (meditation, brain retraining, etc).
Again the ‘mental’ solution part does not mean at all that any of us is crazy neither that this is all in our head.
Like it !!!
I think you laid it out quite well 🙂 Some weakness in the mitochondria that predated the infection I think is the idea.
Cort,
You know what. I am an aerospace engineer and it is probably professional training but I really need to understand things in life. The more I understand things in life, the more I feel empowered and in control of the situation. It is exactly the same thing with ME/CFS for me. The more I understand the potential causes, the more relax I feel, in spite of pretty tough periods evidently, and the less severe the situation is for me globally.
Mind therapy doesn’t work for ME/CFS, many of us have tried it and had no improvement. In fact I deteriorated even though I was doing very good relaxation exercises, body scanning, vagus nerve relaxation etc.
Note: i haven’t had and don’t even have anxiety. I’m a chill person.
I think you are confusing ‘stress’ as the psychological form.
When there’s stress in biology it means things in the environment like food, compounds, toxins, viruses bacteria fungi, exercise, immune responses, hormonal changes etc, that all put a stress cells and cellular signalling. These cells as Dr Naviaux has stated many times are locked in a Cell Danger Response that normally only turns on during infection or injury. For some reason it remains locked on.
There’s been decades of nonsense about the mind therapies for ME/CFS. If it worked I’d be cured and everyone else would be cured now too. But it’s only a few improved who i believe either had recovered and we’re deconditioned, so can pull out if that/ or had psychological fatigue that improved with mind therapy.
Us (the ones with actual ME/CFS) have a biological disease.
The cure will be biological too. Not psychological, not ‘dualism’, just biological.
Like other diseases the mind only helps cope with living with the disease. It doesn’t improve it. If you look at studies on cancer outcomes with will power and optimism, they made no difference to the outcome of these physical diseases.
All mind therapy does is help reduce the emotional suffering of having to deal with such a vile physical disease. I support it being used for only that
I’m truly sorry that you got worse. That said I’m really disappointed with part of the community discrediting other people’s experiences.
If someone says that mental approaches were a part of their recovery or improvement, don’t minimize that just because it doesn’t work for everyone!
I’m not saying mental approaches are a cure-all for ME, they obviously aren’t. But we aren’t doing the field any good if we dismiss the cases where it helped by just saying they didn’t have ME in the first place. There may well be different subtypes of ME and each trajectory is so different due to genetics and all sorts of environmental factors before and after developing the disease.
We should be interested in WHY it helps for some and not for others. Then we could better figure out how to help each and every patient individually. And especially, we would finally fix this divide in the community, which does nothing for it.
I believe a component of ME/CFS impacts the central nervous system. I also believe that mental stress is a trigger for a crash and puts extra burden on the body. For instance, on vacation my ability to exercise and physically exert myself without a crash increases dramatically. For me, meditation and relaxation helps dramatically. I’m normally a pretty chill person, not particularly stressed or nervous, or so I thought, but a few minutes of meditation before bed helps me sleep better and seems to energize my body the next day. Breathing techniques help me fall back to sleep when I wake up feeling wide awake at 3am, which occurs almost every night. I call feel the breathing and relaxation calming the fight or flight response my body seems to be going through.
As an aside, I was diagnosed with ME/CFS by Dr. Systrom who also gave me a small fiber neuro test – positive.
It seems these techniques don’t help everyone, but some might find relief.
Well said, Nuria. Thank you. It astounds me that people who have suffered the pain of being discounted, discredited, gaslit, and judged feel no hesitation to deliver that same harm to fellow patients. There is no single option that helps everyone. Even something as basic as pacing isn’t helpful to all. Neural approaches are NOT appropriate or useful for everyone but for those they are, they can be a game changer. My point, though, is not to promote neural approaches. Instead, it is to promote change in how we treat each other. It is helpful to share what works and doesn’t work for us so we can learn from each other and decide what to implement based on what is appropriate for our individual circumstances. It is harmful to discredit, discount, gaslight and judge others because their experiences do not match our own. We are not all the same. Mutual support and respect will go a long way to healing the harm that we’ve all experienced. If we aren’t willing to do that among ourselves, how can we expect it to come from others?
I said “… it’s only a few improved who I believe either had recovered and were deconditioned…” (what I mean here is they genuinely had ME/CFS but their bodies recovered (i.e. PEM had previously gone without them knowing). But because they were so weak from years of being unwell they remain deconditioned. People could be in that state for many months. (It happened to a friend of mine).
Then they use mind therapy and start finding they are able to push through. That’s because there was no PEM to block their recovery).
That is exactly happened to Prof Paul Garner. Back on the 5th October 2020 He replied to my email, to tell me he “didn’t have problems with pushing through” and was “doing 5km daily walks”
Note: That was before mind therapy. Yet he now he is on a campaign falsely claiming it was mind therapy that cured him.
The important issue here is he and others whose PEM had ‘gone prior’ will recover with mind therapies. But if you have severe PEM after exertion then using their techniques a crash will occur.
If someone has a psychological problem on top of ME/CFS then yes that will help tone down exacerbation of symptoms. I think it’s fine to do meditation and other stress relief exercises if a person is constantly on edge as is energy consuming so reducing that energy use for them can reduce a flare. but my comment was in reply to someone saying that to do mind therapy with medication will help people recover. I’m saying that’s not true and there’s no evidence of that either.
I am talking about people who don’t have mental illness but do have ME/CFS. It’s not true that it’s rare. Some people with ME/CFS don’t have anxiety and some do. You lot are adding a ‘psychological comorbidity’ onto ME/CFS. Which is a separate issue.
This is a biological disease, not a psychological one.
For decades those with mental illness induced fatigue ‘without PEM’ ended up in ME/CFS clinical trials due to misdiagnosis, and that data muddied the waters in the results. Which is a big reason why this disease hasn’t progressed in biological research. And is because it was suggest that ME/CFS was psychological, was a huge reason why government funders have have shied away from biomedical research.
It’s also a reason why the psychiatric community (Simon Wessely and his goon squad) hijacked the disease with their fake GET and gaslighting CBT mind therapy to challenge one’s sickness belief nonsense. That therapy actually harmed people.
There is so much to unpack here, I don’t know where to begin and I’m not going to try other than to say quite emphatically that neural approaches do NOT, and let me say that again, do NOT suggest ME is psychological or a mental illness or a psychological comorbidity. Neural retraining goes well beyond CBT or GET or meditation and is very much related to biology. Autonomic nervous system dysregulation, which is biological and which neural approaches help regulate, is not being ‘anxious’ or having a mental illness that ‘muddies the waters’. Those of us using these approaches are as educated, experienced and knowledgeable about ME as everyone else. We are not confused about PEM and deconditioning. We know what helps us and what doesn’t. Improvement comes in different ways for different people. Can we not accept and respect that?
Hi Michele,
I have learned for a very long time in my little career (I am just a not very educated and knowledgeable aerospace engineer who can not understand complex concepts because I am working in such an easy field) that it is very hard and not very useful either to argue with people with such a rigid attitude.
Indeed, they have a hard time to understand the concept of discussion : to exchange ideas on a open mind basis in order to end up with a better comprehension of a situation.
Generally, they just want to have the last word because THEY KNOW. I am always very wary of people who know because they tend to jump to conclusion before the full job is completed.
By the way, in case this could be useful to someone, anxiety needs to meet very very specific criteria of circumstances and severity to be diagnosed as a ”mental illness” in the DSM5-TR.
You are so right and I only occasionally engage on this topic because it is so fraught with misinformation, disinformation and emotion. With what recently happened to Dr. Eleanor Stein, though, I am less inclined to say nothing. Doing so is giving a group of anti-neural training people a louder voice than what is healthy for the community. It is causing harm.
https://www.eleanorsteinmd.ca/blog/canmebecuredwithneuroplasticity
As I’ve said elsewhere in this blog and in other places, I am not here promoting neural training. While it can be very helpful, itis not a cure. It is not appropriate for everyone. As with every other option for ME/LC, results are mixed.
What I am calling for is for us to come together as a community. To listen to each other and respect that we are all different in what works and what doesn’t. Our individual experiences are valid and deserve respect. At the same time, none of us has the right to claim our version of ME is the only right version or that what works or doesn’t work for us applies to everyone else. I’m going to continue to call for mutual respect because I believe it is long overdue and essential for our health.
If you really explore these practices I think you’ll find that they’ve focused on altering the nervous system – in particular the sympathetic nervous system – very different from psychology.
Do you have a link to a free video or website on how to alter the nervous system with brain training? With the steps on how patients can do it. As all the links I can find have a very high prohibitive cost paywall,
I did the expensive Lighting Process and it was pure gaslighting by making me ignore my symptoms, ignore I had a physical disease, which triggered a severe crash.
I tried NLP too which was more of telling me to ignore symptoms. Both ignore we have PEM
If there’s a free one I’m sure many of us would try it.
Thank you
I have not tried brain retraining methods directly yet but I have tried basic in-depth breathing method (abdominal breathing) and I am actually trying meditation ”MBSR by John Kabat-Zinn”.
What I understood from all these methods is that the goal was to help calm down the sympathetic system (accelerator pedal) and leave place to the parasympathetic system (break pedal) in order to restore homeostasis (equilibrium).
I am fully convinced like you that ME/CFS is a biological illness and I have never really been an ”haaauuum type of guy with parsil in my ears”. The original image I had from meditation was a weird guy levitating above a nail carpet in one of Tintin’s album so you see where I am coming from… Then, I never really believe that this would do any miracle and it did not either so far.
These method seems to be effective at what they claim, remaining more calm and cope better with everyday stress of life as well as return more rapidly to a calm state if you ever lose it a bit. Sleep was alos improved by a colateral effect of being globally more calm.
Except that, which is already pretty good I find because it costs nothing except the price of a book or two, it never did anything close to cure my ME/CFS even with a 100 foot pole.
I still have severe PEM when doing sport once a week and it still takes me the whole week to recuperate. Nevertheless, I do not want to stop completely: being a sportsman born, I would otherwise feel like a living dead.
By the way, if the methods you tried so far were really meant to make you believe to ignore your PEM symptoms then these methods were garbage in my opinion and I can understand why you are so angry at them. I guess and hope they are not all the same.
Hope your new trial will be succesful if you ever attempt it…
Hi Canada, glad to hear you benefit from the MBSR programme. I am a long time follower of the path of meditation, MBSR, and Buddhism and I also benefit greatly from it in my recovery.
I think that the key to benefit from this practices is not to practice them as therapy but without attachment to a specific outcome.
That’s why I am extremely wary against folks who advocate for meditation as a therapy in ME/CFS and everything else!
A personal feedback: For me, exercise has become another pillar of my recovery, and I actually do use it as a therapy. However, I do it daily in small doses in order to never induce PEM and an ME flare-up.
I want to urgently let you know that you should try the same. Because when you exercise and go over the limit of your PEM-threshold you don’t allow your organism to stabilise. With every inflammatory flare-up toxins are produced that damage the mitochondria, the vessels ect. and thus hinder these tissues to regenerate. Also, whatever it is that keeps ME going has the best conditions when you practice this life-style.
If you want to recover the most important thing in ME/CFS is to avoid flare-ups. A British physio therapist working for the ME association has produced the best instruction to pacing and exercise.
After 30 months with this rehab approach I am actually back at spinning and running for 30 min, being able to do more weightlifting, and following a one hour intermediate class.
My experience is that the longer that I have managed to avoid PEM the more far away I am from producing a flare-up. Which means I can slowly do more and more and more without triggering PEM!
Think about it.
https://meassociation.org.uk/literature/items/pacing-activity-and-energy-management/
The link to the document on pacing.
HI Lina,
I am pacing all the time, every day for everything. This is the only thing that works for me plus a bit of MBSR.
I am hypersensitive to any supplement I may try. Even with the go very low and very slow approach, I always end-up with all the side effects without almost any benefit. I become cranked like hell like in the movie with Jason Statham and then I absolutely have to stop because I can not sleep at all anymore. Sleep deprivation is no good for mental health, as you know.
Regarding sport, I can not do it every day even just a little because it severely affects my sleep. Then the best solution I found for me is to train only once a week and then I have 3 days to recuperate to about 85 % (very long nights with many naps because I work 4 days a week and the remaining 15 % is back during the rest of the week.
This is this way for me or no sport at all. Sport is my mental equilibrium, my way of deconnecting from work and hard thinking (aerospace engineer). Without sport, it is not worth living for me…
Cheers
Hi Lina,
I answered you rapidly the other day (back from work and tired).
More precisely, I am using exactly your strategy of a bit of sport very regularly (without never exceeding my PEM treshold) almost all year long during fall, winter and spring but not during summer.
During my younger years 15 – 20, I was an alpine ski instructor and I was training hard for road bike racing (although I never raced in the end).
Normally with this strategy, without pushing myself at all, I can do about 30 minutes of stationary bike in the morning, about 15 minutes of weight lifting (leg extension and flexion machine) in the afternoon and about 15 minutes of other core strength and stretching exercises in the evening a couple of times a week.
During winter, I can also do one day of alpine sking with only a pretty light PEM. However, during summer, I only do road bike and I give up everything else because it is too demanding. Even with only one ride a week I have strong PEM but I have no fun looking at the birds on my bike. I need to train so this is more demanding and I have to dig in my battery.
Yes I’ve done similar for many months at a time. Like Mindfulness, Yoga Nidra, also gentle diaphragmatic breathing exercise exercises. Boxed breathing, mini breath holds, all sorts of breath methods.
As mentioned earlier I’ve done LP (that’s the worst) also NLP, also a friend paid for Gupta (brain training) and shared with me the methods.
Yet I continued to deteriorate with ME/CFS. My friend stayed the same. We did hear of someone improving though.
I see ME Action have a page on Gupta who got in trouble by the Advertising Standards Authority for making false claims about it’s effectiveness
https://me-pedia.org/wiki/Gupta_program
I do however still enjoy doing these different methods for simple wellbeing. As is the closest to a high but is natural.
Especially the body scan and visualisation meditation as i can feel the parasympathetic/sympathetic nervous system actually clicking over during it. So is a good way to get to sleep or back to sleep.
Unfortunately hasn’t improved the disease
Pretty much the same thing for me so far. Anyway, I do not really expect more from these methods either. I expect a real effective medication ASAP.
This disease is a life ruining event when it starts. The worse aspect for me: you know that special list of expectations in your life when you are in your 20’s, the to do list or the bucket list, whatever it is called. Every thing goes overboard one after the other and none of these elements occurs as originally planned.
A wife or a girl friend, kids, many friends, gone in a second… When you can never do anything and refuse almost every activity or invitation from everybody because you are too tired, they stop to invite you and/or simply dump you. This is tough mentally to deal with all that. Anyway, I am sure that you already know all that.
Never give up is my way of thinking but I am more running on my will than on my body.
I have taken 5 infusions of NAD. My Myalgic Encephalomyelitis has been remission for 6 weeks so far. I feel like a new person. mitochondria and they are depleted of NAD+
Thank you for sharing what’s working for you!!!
No, I am not confused about the stress definition. I perfectly know the difference between biological stress and psychological stress.
I fully agree with the explanations of cells being latched on in an emergency or danger state. Of course ME/CFS is a biological problem and any relaxation technique, brain retraining or whatever you want to call them are absolutely no cure to this physical disease.
I am not saying at all that psychological stress is the cause of ME/CFS. I am only saying that that emotional stress, if you have some, induces a second vicious circle layer to the base physical problem and then, the more relax you are in your life, the better you are able to cope with the base physical problem. We are saying exactly the same thing.
Good for you if you are a chill person and if you never had any anxiety related to this disease (you are a very rare and lucky one because I personally find this disease extremely stressful on all aspects of my life). I am also very surprised that you deteriorated because being more relax, even if you already are, can normally do no harm but this is your personal experience with these techniques, so be it.
Hi Canada; I reply here because there is no button anymore above…
Interesting to hear about your health level and what you are still or again able to do. That’s not bad.
On the other hand you of course have losses too. I am sorry that you think that it is not possible for you to have a girl friend or a family anymore. It seems that’s something important for you.
Regarding the physiotherapeutic question. What I see definitely different from you is that I think of myself as in recovery and my most important goal is have a full recovery if possible.
My doctor has told me that he has seen it happening for some of his patients in ME/CFS even when they had been severely ill (moderate to severe) for some time. He said that they had their old lifes back but added that he had also seen that such patients could be well for several years and then in a demanding life situation still could have an ME flare-up.
As I said it is my theory and I think it is supported by what clinicians say, by provoking PEM and flare-ups we disturb the organisms capacity to heal.
Since I believe that HHV-6b is at the core of ME/CFS pathomechanism and based on my observations of fluctuation and quality of my symptoms, my theory is that light PEM and light flare-ups equal slight HHV-6b reactivation in the cells they are most likely to break out, the immune systems T-cells (and from my own observation possibly also endothelial cells).
If this is correct it means that every flare-up weakens the immune system (and vessels) a little bit. And when you do it on a regular basis then you induce that damaging process again and again. I wouldn’t like to do that.
I prefer to go more slowly but hopefully farther in my recovery in the long run!
Hi Lina,
I think this strategy of avoiding to exceed the PEM treshold and then any flare-up is perfectly logical. This is my experience too.
After so many years dealing with it now, I have found a kind of equilibrium which allows me to avoid flare-ups almost all year long but during summer. Indeed, summer is the period of the year which is the toughest for me and I guess it is precisely for the reason you mentionned: I am pushing to hard. I know it but I keep about 4-6 weeks very quiet at the end of the bike season and I can then fully recuperate until next season. Not ideal but it allows me to still enjoy bike.
Not sure however if this really prevents me from getting fully rid of ME/CFS because I have already spent a couple of summers just looking at the birds on my bike (not training) or without bike at all and it did not lead to that perfect conclusion.
Regarding the root cause of ME. After the latest papers from Cort, I like the story being built actually.
We know for sure that mitochondria are not working properly. It looks like ME/CFS could start with the presence of a genetic predisposition (kind of mutation) and could be triggered by a stressor that would induce a kind of latched on emergency or panic state in the cells. I have read that EBV would be a prerequisite trigger for the development of Multiple Sclerosis. Then the assumption of HHV-6 playing an important role in ME/CFS as well is very serious. What is still not clear I guess is if the mitochondria are broken forever by this stressor or simply affected in their function while the stressor is active. I have read that this could be reversible.
Sooner than later, we will understand what is going on. After that half the job would be done. We could then concentrate on the development of a proper medication or still better the identification of an already existing one.
Meanwhile, pacing and MBSR are helping a lot to deal with ME/CFS for both the body and mental aspects.
Cheers
I’ll reply within the next days. For the mitochondria you are maybe interested to have a look at that research summary by the British ME Association:
https://meassociation.org.uk/2019/07/mea-summary-review-the-role-of-mitochondria-in-me-cfs-13-july-2019/
I can’t judge the discussion right now because I have to read up. But judging from their documentations aimed at patients to help them help themselves they’re the leaders in ME/CFS in my view. So when they say that the research has already shown that the problem does not start in the mitochondria but “upstream” I sounds credible to me.
In fact, B Rob, many of us HAVE improved with neural therapy. To date, there is no cure for ME. There are, though, different strategies and different treatments that lead to improvement in some, but not all, people. Neural therapy is one of many options that may or may not lead to improvement. Discrediting the experience of others is not helpful. In fact, it is harmful to all of us.
Show the scientific evidence with Peer reviewed Randomised Controlled Trials. And studies that were able to repeat the same outcome
It would be wonderful to have this kind of evidence for every treatment or approach for ME. We can only hope that someday it is the norm. Sadly, evidence like this is in very short supply and is very expensive to achieve. If we condemn everything that isn’t supported by repeatable peer reviewed RCTs, our list of options will be very limited. I don’t personally know of any patients who are willing to do nothing to help themselves while science takes it’s time to catch up to us. Can we not show respect for people who have experiences different from our own?
There’s millions of people in the world with ME/CFS and Long Covid. Neural brain training has been around a long time now. If worked beyond placebo we’d see tens of thousands of patients recovered or vastly improved by it. We don’t
I’ve been to these brain training websites and read the testimonials, and I see only a few dozen people claiming it worked. Even if it was a few hundred people that’s still so tiny compared to the millions with ME/CFS, or thousands that have tried it.
I’ve seen far more people in online forums and social media saying it didn’t work than did.
If it works then why aren’t there any decent studies showing it works? I can’t find any. Yet they charge big bucks to do the course, lots of money being made yet not put into proving it works. Thats a red flag. I also see the practitioners of these courses coming into online forums as bunched claiming it works, but of course they will as they are motivated by money.
I can accept that people who did recover actually were on the cusp of improving anyway because there’s plenty of recovery without mind therapy. Had they tried a certain supplement at the time they recovered they’d be adamant it was that specific supplement.
I’m not saying to people don’t try it. I did. I’ve tried so many different things.
On the issue of money, why are these brain training therapies all behind paywalls? Surely if mind therapy worked there’d be someone so excited to guide people in a YouTube video for free.
Virtually every trade and skill is shown in ‘How To’ videos by someone on YouTube for free. If you have a link to a video on it id be interested to see if it’s similar to what I did. And if you are sure it works you will be helping others at the same. Or do they have to pay?
Fluge, Mella and Tronstad (very biomed oriented people) might be able to explain why some patients benefit from mind-body techniques:
«In our model, clinical symptoms of ME/CFS are related primarily to the inadequate autoregulation of blood flow yielding tissue hypoxia on exertion, but are also influenced by the compensatory adaptations from increased sympathetic output and from metabolic shifts.
We speculate that cognitive techniques, which are reported to help subgroups of patients, might act by modulating the sympathetic output. If so, one would expect a greater benefit for patients with less ongoing immune activation and less vascular dysregulation, but with main symptom contributions from the secondary autonomic adaptations.
Conversely, patients with active immune disturbance and ongoing vascular dysregulation as the main symptom generators would have less impact from cognitive intervention, although psychosocial support and coping strategies may still have a beneficial impact on their quality of life.»
Ir the mitochondria has less oxygen, it will die more often, regatdless of the mutations. If some of the lack of oxygen is caused by an over-active SNS, calming the SNS through e.g. mind-body could in theory be beneficial. For some, that’s enough to get better. For others (probably most), it’s not enough.
Thanks! That’s in line with what I’ve been proposing for years. 🙂
Fluge, Mella and Tronsta Had a massive difference between their first trials compared to their big RCT trial. It was an utter flop. The last trial was so profoundly different I suspect they manipulated the data in the first ones to get funding.
I have no trust in those researchers whatsoever
@B Rob
It’s completely normal to have different results between phase 1, 2 and or 3. Probably more normal than not – most drugs don’t make it to phase 4. That’s also why they stopped, because they trusted the results and want to focus on finding an actual cure. How is that a bad thing?
They also work with Ron Davis’ ME/CFS Collaborative Research Center at Stanford University. If Davis trusts them, I’m inclined to do so as well.
Furthermore, Fluge and Mella (I’m not sure about Tronstad), were cancer researchers before they stumbled upon an ME patient that fully recovered due to chemo. They mostly left the most lucrative and prestigious field for ME – the exact opposite.
They are known for doing a lot with very little funding, and they are currently trying to develop a more gentle way of measuring PEM (text in Norwegian, but you can see the method they propose): https://www.facebook.com/100064524226847/posts/pfbid0ihusHZ1w24Ybc1s2YQ6LpbzTpafxVBoiuNZ3yQPFSVJYu4UUsbzjKQZFG7ALMgiBl/?app=fbl
You are on a roll, sir!
Please do not impugn the (few) researchers that are devoting their time and energy to us…We try not to do that here!
You might remember the Daichi Sankyo Miragablin trial. Daichi clearly thought it was a can’t miss drug and put on a worldwide 300 clinical site fibromyalgia trial which totally flopped! These things happen.
https://www.healthrising.org/blog/2017/07/08/mirogabalin-fibromyalgia-drug-fails/
The same thing happened with Tonix – which later managed to turn things around when they changed the dose. The point is that the early studies suggested they had the right dose – but they were wrong.
Carmen Scheibenbogen believes that the maintenance dose in the Rituximab trial may have been too low, by the way.
I am with Rob. I have said it before. The scientific evidence to claim that mind-body practices are a good support for everyone’s well-being whether ill or not with whatever disease was already brought foward back in the 1970ies and 80ies!
And I have experienced it myself first in my life before ME/CFS when I struggled with anxiety and depression and now with ME.
Therefore the research that is presented here is superfluous. Even when it is done with merely speculative ideas about what’s going on in ME/CFS. ; )
I agree that many people who have tried mind/body approaches have not met with success – but for me this goes too far: “Mind therapy doesn’t work for ME/CFS, many of us have tried it and had no improvement. ” We’ve had lots of people who said they at least received some benefit from it.
For me, I was one of these people who were highly critically of the mind/body approach. I considered it not only a not very helpful tool (beyond normal stress reduction, healthy for everyone) but I did find its promotion to be counter productive.
First of all because ME is not in the mind. But more so because I feared that (poorly designed) studies looking into the benefit of eg brain training for ME would take money away from needed biomedical research and would serve as a cheap cop out for insurances and other providers.
I still see this danger. With LC we see that big insurance companies in Europe are conducting studies that “prove” that exercise and mindfulness can cure LC patients. The ones not cured have nothing in their hands to claim social security etc.
That said, I began incorporating regular mind body techniques this year as my health has dramatically declined and I really saw a lot of benefits (no cure or drastic improvement). One of the most important and often overlooked ones: Mindfulness helps me with pacing!
I am glad that you could give mindfulness another try. Even when there is so much bad stuff going on around marketing it as a cure in ME/CFS.
It is also a big support for me. The key is to not use it as a cure. Because letting go of needing to be healed comes as such a relief that it is a booster for recovery. That’s why mindfulness works and for the same reason it stops working in clinical settings.
I can’t quite follow you on the idea that it is overlooked that mindfulness helps with pacing. This is one of the greatest benefits. People who actually practice can’t overlook it I guess.
Thank you for your reply, Lina. I think you right about not thinking of mindfulness as a cure. Interesting point about it not working in a clinical setting, that is exactly my experience.
I think my point about mindfulness is, that it is promoted as a cure and many people want it to be one (understandable) and people who are aware that it is not a cure, then reject it completely.
I think of it as a tool for pacing (our most important tool). I think you are right, when you actually start practicing mindfulness you can‘t overlook the benefits for pacing 🙂
Hi Ana, yes I have heard stories of people in clinlical psychiatric settings who were kind of oppressed with mindfulness training. Of course you can teach it differently but the world of the physicians is just very objectifiying and subjecting and thought to offer cures that mindfulness programmes tought by the same people can’t be different from that way of relating to people.
But actually, the MBSR programme was developed in a kind of clinical setting. But Jon Kabat-Zinn asked that the uncarable patients would be sent to him.
Here is an intersting talk of his where he offers insights into the beginnings of his work:
https://www.youtube.com/watch?v=kShpURJOpeE&t=5372s
He talks of mindfulness of inducing healing but not offering a cure.
I wish you joy and insight with your practice!
Did you read the part that explained that fatigue can be generated by the brain? And how they found that Small fiber neuropathy (A common comobordity of MECFS) can be also caused by stress on the brain?
It doesn’t mean that the disease is psychological but that mental stress can be the detonator with real biological consequences.
If all of this is true, it makes sense to me that some people are cured after brain retraining.
ME is not only generated in the brain. Dr David Systrom found impaired oxygen extraction in the muscle cells. He’s currently doing a muscle biopsy study that will hopefully determine why. But he currently believes that the mitochondria are not doing as they should.
Using brain training won’t undo that.
There is a second ME/CFS sub group who don’t have oxygen extraction issues. Maybe they are the ones that could benefit from brain training. But like B Rob said why isn’t there any free information on how to do it? That sounds alarm bells to me
Our organization sponsors the National Birth Defect Registry and we collect data on health, genetic, prenatal and preconceptual exposures in the children of both parents.
Our data include structural birth defects like spina bifida and cleft palate as well as functional birth defects that include immune, endocrine and neurodevelopmental problems like autism.
The online questionnaire for the registry which was developed by a team of scientists has hundreds of questions, but a place to write in a condition or exposure not currently on the survey. We currently have have nearly 6700 cases in the online registry and another 3-4000 in the original booklet format.
We were surprised to have over 539 cases of ME/CFS reported, 422 of these in the children of Vietnam veteran fathers exposed to Agent Orange.
Our hypothesis on the conditions that are increased in the children of Vietnam veterans is that dioxin (toxic component of Agent Orange) has a negative impact on the development fetal immune system making the children vulnerable to autoimmune disorders, cancers, immunological, neurodevelopment problems and more.
https://birthdefects.org/wp-content/uploads/2024/07/Vietnam_Veterans_2024.pdf
There are many other exposures that could also impact fetal immunity.
Maybe we are not going back far enough to understand who is most vulnerable to ME/CFS and Long Covid. Since World War II and the splitting of the chlorine atom, there has been a proliferation of new chemicals in our environment.
Hi Betty,
Very interesting data. Of course chemical pollutants of all kinds may well be a cause of genetic dammages. Indeed, with time going by, I guess we will understand more and more that genetic dammages are not a purely random effect but rather often related to human causes (Irene Brokovich story, etc). AI will probably help a lot soon to investigate such huge databanks. As anyone so far (doctors, research centers, etc) requested your databank for in-depth investigation ?
Dear Canada and Terry,
First, Terry thank you for your kind comments.
Canada, rather than ramble on about what we have done with our data, I thought you might want to read this report about the registry and the scientists involved.
https://birthdefects.org/wp-content/uploads/2024/06/Registry-doc.pdf
Our data have been presented to government agencies, congress and in national media forums.
Thank you for your extraordinary work!
Looking forward to Part 3 – treatment suggestions.
Where there’s smoke there’s fire. Clearly something is wrong with the mitochondria. But are those problems contributing significantly to symptomology, or not? Or are the mitochondria just unfortunate ‘collateral’?
I suspect’chicken and egg’ – a feedback loop. Will effectively treating the mitochondria effectively address the feedback loop?
Is rapamycin part of the answer?
Is viral reactivation the issue? Might PolyBio’s antiviral clinical trial address the mitochondria by addressing viral reactivation?
Praying for meaningful treatments
We have a lot of arrows pointing at the mitochondria, and in some ways we are in the same fix we have always been in – lots of small studies. Hopefully, the smaller studies will add up and we’ll get some nice big ones.
After careful consideration of all the studies that were available at the time my son got sick with EBV-CFS in 2020, I focused on the mitochondria and had him take a full complement of mitochondrial supplements, with the aim of reducing PEM in particular. I believe treating the mitochondria (using PEM as the measure of success) allows the body to recover, allowing the use of neural therapies to seal the deal and the individual to recover to almost normal, as others in this thread have suggested. Interestingly, one of the therapies that helped was low dose naltrexone, which has since been hypothesised to be of use to mitochondria. The mechanism proposed is calcium ion channels, which are within the mitochondria. https://pmc.ncbi.nlm.nih.gov/articles/PMC8313851/
My son recovered after 18 months of fatigue and is living a normal life now. His supplement/medication regimen was D-ribose, CoQ10, Vitamin D/K, curcumin, LDN, methyl B12/Folate, quercetin, high dose B1, magnesium, melatonin, fish oil and methylphenidate. We also tried a number of other supplements with no or little effect. Other things that helped were pacing, Mickel therapy (a neural approach that emphasises doing what one intuits is right based on the messages one’s body is sending) and the Covid-19 vaccine. I hope my little contribution synthesises the approach to treatment of someone here reading this. Good luck to all!
Congratulations and thanks for sharing 🙂 Very good you got after your sons illness quickly. I imagine that helped.
Thank you for the specific recovery details Chris.
So, I guess this explains why you get this from your mother?
If you’re in the heritable group it could explain that. Of course, there may be other things going on. Nobody else in my family has had ME/CFS – including my twin (!) – but my mother did have a lengthy autoimmune illness
Wow Cort, that’s very interesting. I am sure it’s more complex than that but when we look at celiac 30% of the population have the gene and only 2% have celiac. So almost always there has to be a trigger. So maybe even when mothers have ME that doesn’t necessarily mean much?
If the mitochondria is the “energy” part of the cell, and if it is not functioning as it should (or not at all) and if this ‘faulty mitochondria causes all the parts of the body not to function as it should; or ‘slower’ than it should, then it makes perfect sense that the brain, the muscles and even the gut would be ‘fatigued “ or work extremely slow i.e. brain fog. I say this because I had shingles at the same time I had a 9 hour surgery on my spine in 1997. Although I had undergone 6 previous long spine surgeries since the age of 15; this one was different. Since that surgery in 1997, I immediately knew something was terribly wrong. I knew what post operative pain felt like-but this was different. Since that surgery, I have experienced excruciating pain 24/7 even with opioids. I have fatigue that caused me not to be able to take care of my younger children. I have had two different types of cancer, undergoing surgeries, chemo, radiation, etc. I also continued to have ‘shingles’ break out on my stomach and around to my back anytime I am under extreme stress. Interesting, my digestion is so slow that I only go to the bathroom every 10-12 days. Still, I must use an enema to empty my bowels. I have tried many prescriptions to help me have a bowel movement, but it only results in the most intense cramping and stabbing pain but does not end with the desired results! Not sure if this has anything to do with the studies on mitochondria, but I would be interested to know the story of other ME/CFS patients and how their illness started and did it progressed ? Or did their symptoms stay the same? How many patients have had times of ‘remission’? How many suffered with severe ME/CFS and now are free from all symptoms?
I might add that I have been in therapy for a long time due to growing up in a very dysfunctional family. Abuse that I won’t explain here~but it was severe and my case study has been written about in 2 books from 2 Therapists who have treated me. I was diagnosed with severe PTSD, childhood trauma and serious neglect; physical and ‘other’ abuse as well as having a mother in and out of Psychiatric hospitals and a father who would leave me (at 5 years old) at home to care for my 14 mo old brother every day while he went to work and my mother was hospitalized; leaving no food, formula or instructions. My Oncologist found it odd that my extensive bloodwork done before any cancer treatment showed that my cortisol was as low as he (or the Endocrinologist) had ever seen. My Psychiatrist said it was probably due to my adrenal gland putting out so much cortisol for so long during these ‘fight or flight’ episodes , that it just doesn’t work.
I wish all ME/CFS patients were given extensive health histories to be filled out and all sent to ONE research group. Finding out all the symptoms, personal histories, etc. might open the eyes of the researchers as well as us, the patients. Sadly, I still deal with physicians who have no clue about ME/CFS. When I tell them what the long name is~ I always get “oh. We ALL have chronic fatigue these days. You should just get more exercise and good sleep”. Ugh! Why are we STILL getting these responses? I’ve had this disease since 1997. It took 12 years, 61 physicians in 8 States before I got a diagnosis! And yet nothing has changed.
This would go some way to answering the oft ignored issue of wherefore post-exertional malaise?
I would think so…The more stress put on the mitochondria the more they break down. I could see an ioxidative stress cascade that results from that as the “fire” feeds on itself. There are also the studies showing that the body at a molecular levels is just not responding to exercise – because the mitochondria have gotten whacked (?) – and muscle repair mechanisms are not kicking in either.
I’m tired of these studies. More, blah, blah, blah..some contradicting, each other… I don’t what..l’m not a phd
I mean want..
Has anyone tried the ginseng supplement HRG80?
https://pmc.ncbi.nlm.nih.gov/articles/PMC8777686/
Ginseng made me nervous..too much energy, and for me, didn’t seem like the good kind of energy.
But try it if that’s an instinctive thing to do, and let us know. Maybe it will work fine for you. Not meaning to dissuade..
Take care everyone!
Yes with no benefit. But I have been feeling better drinking hydrogen water (not hydrogen capsules).
May I ask what symptom benefits from drinking this?
The symptoms of this illness vary so much from person to person, it’s hard to know what might work best.
By far the main symptom for my daughter is mental fatigue and lethargy. Trying to find things that might help that a bit.
Increased energy, less weakness in my legs. Just a better feeling of overall wellness. I started out low and built up to 3 bottles a day (company recommends 5 per day). At first drinking before bed kept me up but now that I’ve adjusted, it doesn’t mess with my sleep. The biggest help for me (which is also proving to me it is helping) is I am no longer getting any (genetic) Hailey Hailey Disease flares. When my cfs symptoms started, my genetic HHD worsened. I’ve found over the years anything that is mitochondria focused, also helped this genetic mutation. For me it feels like a two for one win. My skin is healed and my strength is slowly coming back. It’s only been 4 months so far. I’ve been struggling since 2008.
I really get it Elaine. I also tire of the endless studies that don’t really seem to take things forward much. Yet, at the same time, I do feel that we may finally be on the cusp of some meaningful progress.
I am hopeful that some treatments such as BC007, or rapamycin, might offer meaningful help for a significant number of people. Of course, I am cautious with that hope. We have all been burnt many times by false hope.
I also find some of the new players like PolyBio energising. And Dr Iwasaki at Yale, who PolyBio are collaborating with. We really need that new blood. Some of the old blood have stagnated, without mentioning names, and haven’t delivered on the promise. It’s going to be these young, energetic and funded entities that will get us the answers. Most certainly not the bureaucrats in their massive, slow moving bureaucracies.
🙂 Unfortunately, its science and lots of studies that will prove out in the end. I agree it’s a long road….
In the Gist, 3rd and 5th bullet points include the name White. Should that be Light?
🙂 Yes! Thanks
Stress, in medical terms, is talking about physical demands on the body, not emotional issues, although mental health can have some effect on how we “feel” or experience and interpret our physical symptoms. The best one could hope for in cognitive therapy is learning to cope with the losses caused by ME/CFS, such as anxiety or depression.
I am emotionally healthy and have no history of mental health problems. My symptoms have gradually worsened over time even though I no longer have to push my energy envelope and my life has become a fairly comfortable routine. The PEM is obvious when I push too much, but that’s temporary worsening, not the day to day symptoms that have me far less active than I used to be when life was harder physically and mentally. Is there a mind/body connection? Of course, but it’s not the kind that we can change with magical thinking. I look forward to more mitochondria discoveries.
I was originally diagnosed with adult-onset mitochondrial disease, because I am heterozygous for a few mitochondrial defects. It didn’t explain the severity of the onset though. I had PEM but didn’t understand what it was until 10 years later when I learned of ME/CFS. Because I didn’t know better, I exercised myself into full disability in 6 months, and couldn’t understand how I could catch “the flu” over and over again. My only symptoms from the mitochondrial defects before ME/CFS were mild. I would go hypoglycemic if I ate a large amount of protein at once, and I ran 2miles every other day, but couldn’t run a 5k without bonking. So I carried a candy bar and didn’t run 5ks. Otherwise had a very active life. I think Epstein Barr virus probably triggered me. So I’m believing ME/CFS is an acquired mitochondrial disease. Am interested in treatments, though standard mitochondrial disease treatments have only helped a little.
Chris, can you tell us what the “standard mitochondrial disease treatments” are, specifically? That would be very interestiing to me and I suspect others, as well.
There is a table in this article. https://pmc.ncbi.nlm.nih.gov/articles/PMC4067597/ The two that work best for me, especially reducing muscle pain and cramping, are riboflavin and acetyl-l-carnitine. Acetyl-l-carnitine crosses the blood brain barrier to help with brain symptoms and doesn’t make you smell fishy like the l-carnitine version. I didn’t notice much difference with brain fog. Low dose Abilify helped me a lot with brain fog. Cort said his next article on this topic will include treatments. If you have a genetic defect, you might have to be on a specialized diet, like low fat for a fatty acid oxidation disorder, or low protein or low carb for other specific gene defects.
Thank you, Chris, I will look into this.
Hi Ann,
A good deal of what I address in my posts is based on “standard mitochondrial disease treatments’ but in the context of the big picture and my own experience as a scientist recovering from Long COVID.
About half way through the following post is a link to a peer-reviewed paper on “Feeding Mitochondria”. There is a diagram in my post from this open-access paper that shows (for example) the various vitamins and other molecules used by mitochondria to make ATP.
https://longcovidjourney2wellness.substack.com/p/tackling-mitochondrial-dysfunction
My posts are freely available.
Kind regards,
Mardi Crane-Godreau, PhD
Thank you Mardi,
That is a very interesting diagram, complex and detailed. I am doing almost all of the things suggested there. CoQ10 didn’t help me, though. Caffeine is a surprise. I am histamine intolerant, so coffee is out, but perhaps caffeine pills are in. Thank you for this.
Greetings Ann,
My approach has been based on revitalizing my mitochondria. This is a long term, relatively comprehensive, self-care routine.
Mitochondria need time to heal. As they engage in biogenesis, they are able to repair many of the random type genetic defects that have occurred as the result of aging, viral infections, and inflammatory events involving diet and environment.
A key point: in order to move toward a healthier state, the rate of repair needs to exceed the rate of new damage.
Another point that is central to recovery. As far as I am able to determine, the repair protocol that I lay out works synergistically.
You mention that Q10 did not ‘help you’. What is the context? Are you young enough to be producing high levels of Q10? Were you using a bio-available form? Amounts? Was it the Q10 that did not make a difference or were there other factors slowing down recovery?
Re caffeine, have you tried green tea?
Addressing the requests of some who follow my posts, I wrote a piece called TLC for Mitochondria about a year ago. It’s a summary. There are certainly refinements.
Have a look. Feel free to write or subscribe. My posts are freely available.
https://longcovidjourney2wellness.substack.com/p/long-covid-tlc-for-mitochondria
Kind regards,
Mardi
Thank you for your considered response, Mardi. I understand why you have said what you said because I say the very same things to people who say, “LDN didn’t work for me” or “I had a terrible reaction to LDN.” What happens is entirely dependent on the context, as you say.
I have a detailed response to each of your questions, but I won’t bore you with all of them. Just a few. 😉
I am a 76 year old retired nursing professor in Canada. I have MCAS and hEDS. I can’t take vitamin C of any type because I get almost instant explosive diarrhea. But I eat a half a grapefruit every morning. If I take vitamin E of any type I bleed like a stuck pig (like waking up one morning with one eyeball filled with blood). I can’t drink coffee because it is high in histamine. I tried green tea and my teeth turned a disgusting shade of brown within two days. I will try caffeine pills because I actually like the effect of caffeine.
I do take large doses of K2, D, B complex, melatonin, lyposomal glutathione, enzymatically modified iso-quercetin, magnesium, NAC, luteolin, digestive enzymes, H1 and H2 blockers at triple the recommended daily dose (therefore I have to take HCL so I can at lease digest SOMETHING), methylene blue and low dose psilocybin, naltrexone four times a day, urolithin A, pycnogenol, resveratrol, a brand of “ghost” probiotics (information available by request), and I do 20 minutes of red light therapy twice a day, etc.
Doing all this has helped me a lot. We live on a small acreage and I garden and have chickens. I eat no sugar whatsoever and even limit consumption of honey and maple syrup 🙁 So I consume a lot of stevia and monk fruit extract. My husband makes all our own sourdough bread. We eat grass fed meat from our neighbours and maintain an orchard of different kinds of trees. I am in the midst of fall canning right now. All food is organic and mostly grown by us, except in the winter when I have to buy it. No soda, no candy, no pretzels or any snack, and we drink sheep milk and water. When I splurge, it’s on organic corn chips and bean dip. Whoopie.
You’re right, I probably didn’t give coQ10 a fair try. Sigh. What brand, with what formulation, at what dose, do you recommend?
Hi Ann,
First, so nice to meet you!
I’m a retired professor from Dartmouth Med School… so similar to your background.
I’m also in my late 70’s, live on a small acreage in Vermont, grow our own vegetable (sorry no chickens yet), purchase other food from local farmers who follow organic oriented routines.
While I’m a graduate of Dartmouth Medical School, I have a PhD, and am not a clinician. Thus, I function as a teacher, scientist and writer. When I lay out information, I err on the side of caution. I don’t generally directly ‘prescribe’.Rather, I describe and suggest that people should pay attention to their own bodies, size, age, sensitivities etc.
That said, up until about a year ago I was taking Ubiquinol (a bio-active form of Q10). I then switched over to using MitoQ. My reading of the literature is that the second one may have a slight advantage over the simple Q10. I take their standard dose which is 2 capsules each morning.
Part of my cautious approach is to try things myself in order to feel a sense of confidence before suggest more to others.
One thing that I plan to write a post about is the vitamin-like molecule called PQQ. Rich sources include kiwi fruit, papaya, green peppers and parsley. PQQ supports mitochondrial biogenesis. I see its results in my skin, nails and especially visible is my hair.
If you read my post about bone health I point to the fact that I’ve recently reversed a 20 year decline in bone density. Was it the overall focus on mitochondrial vitality, the PQQ, both, or just good luck? If you read the article on the reported effects of any COVID infection on bone health, my situation seems to be an outlier.
If you act as an advocate for public health issues regarding C-19, I urge you to get more information out about the COVID-Bone connection which incidentally also includes the the gut.
Thanks so much for taking time to write. BTW, I instantly admired you writing style, grammar and spelling. So nice to read a piece where the author cares!
I hope that you will stay in touch.
Kind regards,
Mardi
Hi Ann,
I thought that I’d included the link to the bone health article.
You can find it here:
https://longcovidjourney2wellness.substack.com/p/bone-cells-have-mitochondria-too
I take Q10 (the last blood test shows that I have a deficiency). I also take a complex which includes Vit B, Vit C, Ginseng and guarana. Guarana is one of the foods that contains the most caffeine. I also take iron, Vit D, magnesium, and hydrogenated water. After 5 months of this treatment, I feel better, but I am far from being cured and that does not prevent PME, but on the other hand, these last less long.
Cort you have written a very detailed, complex and overlapping story here. This took a lot of time and effort on your part. This is a superb summary of the research on this topic. I commend you.
You are such a good writer, concise, clear, and understandable. I believe you have gotten better at this over the years, too. When I look at your earlier blogs, they are good but not as good as the recent ones.
This is hard to do and you are an excellent example to all who attempt this kind of summary. Journalists who write for national health publications could learn a lot from you.
You could have a full career somewhere else, but I’m glad you are right here. Cheers.
Ahh! Thanks Ann1. I do think I’m bit more organized with the writing- it seems to be flowing pretty good right now (knock on wood!).
Glad to be here. I count myself very lucky that my bout with ME/CFS left me able to do something that fascinates me 🙂
Dear Cort,
As a longterm reader of this blog, may I second the comment, above, about the incredible way you hold this space.
Also I would like to share my experience of ayahuasca and healing from ME, a year ago. But only if there is an appropriate place or time to do this, so please advise.
Very best wishes
Thanks, Petal – Very interesting about ayahuasca given the studies exploring the use of psychedelics in fibromyalgia that are underway. I will be in touch 🙂
Hi Cort,
Nice job on this important and complex topic!
It seems likely to me that viral-infection-induced mitochondrial DNA (mtDNA) damage is central to ME/CFS as well as Long COVID.
Educating patients about the nature of mitochondrial biogenesis (repair and replacement) and mtDNA damage are first steps. Understanding the relevance of dietary choices and which supplements are vital to mitochondrial function is the next step.
Recovery from Long COVID seems linked to mtDNA biogenesis happening at a more rapid rate than ongoing damage and degradation.
Taking care of our mitochondria is a primary focus of my Substack posts, Insights on a journey back to wellness. Posts are freely available.
https://longcovidjourney2wellness.substack.com/p/long-covid-tlc-for-mitochondria
Thank you again for a really nice job.
Kind regards,
Mardi Crane-Godreau, PhD
So how could the DNA mutations be fixed if it’s even possible? I don’t get it. How can a stressor change your DNA, if that is possible then it should be possible to change it to a correct state, right?
Please, somebody explain it to me, I am not well versed on biological and medical science.
Hi Angel,
We have two types of DNA, nuclear and mitochondrial. The nuclear DNA is inherited from both parents. Mutations to nuclear DNA can generally not be repaired.
Mitochondrial DNA is different. Up to a limit, the mitochondria themselves are able to repair the kinds of mutation that happen from events of living, like poor diet, environmental toxins, and in the case of diseases like CFS and LC, they seem to be able to repair a good deal of the damage that happens from viral infections. (But this requires time and nutrients needed for repair.) The repairable kind of mutations result in Secondary Mitochondrial Dysfunction.
The process of repair and replacement is called mitochondrial biogenesis. It is ongoing, every day of our lives.
One important exception. Some people are born with defects to their mitochondrial DNA. That can cause Primary mitochondrial dysfunction. This type of mutation cannot be repaired by the mitochondrial.
Examples of how stress can cause biological damage includes various stressors that can cause what is called oxidative stress. Poor diet, a lack of protective nutrients, environmental toxins and certainly viral infections can all cause damage. One form of damage is when molecules called reactive oxygen species interact with DNA and cause mutations.
If you’d like to learn more, I suggest a book called Mitochondria and the Future of Medicine. Most of it is pretty readable.
I posted a book review on it here: https://longcovidjourney2wellness.substack.com/p/mitochondria-and-the-future-of-medicine
I hope that this helps.
Kind regards,
Mardi
My disease started after the HPV vaccine on January of this year. Little did I know that MECFS, POTS and Small Fiber Neuropathy were linked to it and that there was already documented.
After spending the last 10 months researching this illness and after reading this post, it made sense to me.
It seems that the vaccine is too strong. So much that the stress induced by it damages the body and the mitochondria which causes all of the problems as pointed on this article.
Small fiber neuropathy being triggered by brain stress blows my mind but it make perfect sense after reading the experiment in the paper.
I just hope there is a way to reverse this. I don’t get how this disease has existed since ever but it’s so unknown and under researched. I also don’t get how drugs well known to be harmful are legal and kept in the market, this should be an incentive to understand this disease as people are always at risk of developing them. It should be unacceptable.
Angel…
It’s called the biggest cover up known to man
The truth is coming out
I think this is why “they” keep changing the name(s)
They called this Yuppie flu in the 70s and the list of names goes on from there
Gulf war illness….the soldiers all got vaccines prior to battle…lots of vaccines
I’ve been vaccine injured my entire life and I’ve been lied to my entire life by the medical mafia
I am sorry it also happened to you. I am also sure there is a cover up, vaccines are a bussines after all.
I just want to get out of this nightmare, I don’t want to live like this forever. There should be something that can be done.
Angel…if you go to the Flccc website there are protocols to try for vaccine injury etc.
They also have a “find a doctor” section. I found it hard to navigate though
But, they are good folks doing good things
Hi everyone
I think it’s great to look at the mitochondria.
What I don’t understand is why there is so little consensus in ME/CFS research about what is established knowledge and what is not.
There is a research summary on ME/CFS and the mitochondria that was published by the ME Association back in 2019 that claims that it is clear from the research that ME is not mitochondrial disease but there are toxins produced by ME/CFS inflammation processes “upstream” – in the blood – that are transported everywhere and that cause damage to the mitochondria.
Then in 2020 Bhupesh Prusty could show that the toxin is mRNA produced in HHV-6b early replication. Viral mRNA is known among infectiologists to trouble mitochondrial processes, possibly because this helps to protect the viral replication from fought effectively by the organism.
https://meassociation.org.uk/2019/07/mea-summary-review-the-role-of-mitochondria-in-me-cfs-13-july-2019/
https://pubmed.ncbi.nlm.nih.gov/32327453/
I sometimes wonder whether the phenomenon and politics of Long Covid have actually not brought the much needed boost to ME/CFS research but have rather derailed it.
Hi, as this is biological. What happens in the brain and body of people that recover or go into longer term remission? What happens to their mitochondrial, metabolic, neuro and immune dysfunction? It simply reverts back to how they use to be?
That seems bizarre. I recently had 2 weeks like that, even my sleep and brain reverted back. Now it was still this new unusual deep sleep but I slept more how I use to. It seems crazy that recovery everything is reversed. Thoughts anyone?
This is a very insightful article on how damaged mitochondria may contribute to chronic fatigue syndrome. The connections between energy production, oxidative stress, and immune dysfunction are becoming clearer thanks to research like this. For those exploring further testing options, you may find the mitochondrial disorder testing services at FML Dubai helpful, as they provide detailed assessments that can support better understanding and management of mitochondrial-related health concerns. Thanks again for sharing such valuable information and keeping this conversation moving forward.
I’m 76 and female. I was homebound from undiagnosed ME/CFS during the late 1980s and to mid-1990s. My daughter has been disabled with diagnosed ME/CFS since 1997. She was extremely active in sports. She got the flu at college and never recovered. We both kept pushing through until we no longer could expend any physical energy without collapse. I consider myself functional, but I live within a specific envelope. The mitochondria aspect rings true for us because my mother also had undiagnosed energy expenditure problems. We think she had fibromyalgia in her later years of life. Always the pain. My sister has diagnosed fibromyalgia. We also all have the body type that is hyper-mobile with the effects of dysautonomia. The younger members had massive mast cell reactions–but not my mother or her mother. So I’d say we are in the “heritable” group of sufferers. What we ALL did that made us worse over years is that not taking steps to rehab when we weren’t feeling well. We all kept going until we could no longer–especially my daughter, who was a dancer/gymnast and use physical activity as a release of stress. We’ve been addressing various issues that seem to have some treatment–the dysautonomia and mast cell activation. I pursued riding a stationary reclining bike after I had to stop working–for two years–until I realized that I had not gained one bit of stamina and was exhausting my leg muscles every day before I could do any home chores. Stopped exercising before that was “a thing.” We know we’re all individuals physically, so I think it’s not surprising that we have different reactions to same treatments. I’ll pretty much settle at this point for any treatment I can get that helps me feel better. And I’d love to see my daughter return to being able to interact with other people the ways and places that most can who are her age. The whole extended family have variations of our differences but most have not become disabled. We do know also that our methylation differences have likely contributed to our family mental health conditions. The complexities seem endless. Thanks, Cort, for once again dissecting what is being done by researchers and giving us some hope.