

Dr. Camille Birch has a PhD in biomedical engineering and hails from the Hudson Alpha Institute for Biotechnology at Huntsville, Alabama. Hudson Alpha, only 11 years old, is one of those new biotechnology efforts that’s using sophisticated bioinformatics to understand how our genes affect our health.

The Hudson Alpha team is doing sophisticated genetic analyses not used in ME/CFS before.
Dr. Liz Worthey and Dr. Birch believed that genetic mutations in ME/CFS might be altering metabolic pathways and causing an “unstable cellular energy state” in ME/CFS. They’re using a variety of algorithms, including one called “custom network analysis”, to examine all sorts of genomic abnormalities (single nucleotide substitutions, structural variants, fusion products, expanded tandem repeats, and variants in regulatory regions). (Yes, our genome is very complex…)
The type of ME/CFS you have, they think, might depend on which metabolic pathways your genetic mutations are whacking. Boy, does it appear that they hit that nail on the head in this study.
 Health Rising’s Quickie Summer Donation Drive is On!
Health Rising’s Quickie Summer Donation Drive is On!It should be noted that the SMCI, which funded this work, did not go to them – this genetics team went to the SMCI. Dr. Birch, who has a family member with ME/CFS, checked out past ME/CFS studies, decided genetics might very well play a role, got the OK to submit her grant proposal to the SMCI – and now a new research team is working in this field.
If you tend to roll your eyes when someone talks about genetics, definitely watch this fascinating webinar.
Genetic Diseases
Dr. Birch pointed out that mutations in just one gene can cause a bogglingly wide variety of illnesses. Mutations in just one gene called lamin-A, for instance, are associated with no less that 11 diseases – many of which have no relationship to each other!

Dr. Camille Birch has a family member with ME/CFS.
On the other end of the spectrum, all 14 subtypes of glycogen storage disease are associated with completely different genes. These genes affect glycogen synthesis, breakdown or in other ways, but all of them end up impacting energy production.
One gene and its genetically modified protein, then, could conceivably be responsible for all the different manifestations of ME/CFS, or, on the other end of the scale, a raft of mutations in different genes could be responsible for, or contribute to, what we know of as ME/CFS.
This study looked at ten patients from Dr. Younger’s cohorts, but what it lacked in size it appeared to make up in depth.
Risky Loci
First they looked at relatively common gene variants or mutations found in the general population and known to have negative effects. They have found 32 of what they called “risk loci” in ME/CFS.
It wasn’t the individual risk loci which stood out, though: it was the pattern present in them. Geneticists always like it when closely aligned genes pop up in their studies. That suggests a problem – perhaps large enough to cause disease – is showing up in one area of the body.
Birch’s sample size was small, and she needed a way to zero in on the potentially relevant genes. When she asked the program if, say, 30% of the general population has a certain variant, what was the probability that a large portion of her ME/CFS group had that variant?
The program told her that it was extremely unlikely that 5 of the 32 loci would be found at such high levels in the ME/CFS group. Bigger studies are clearly needed, but given the probability that these were real findings – that these gene mutations likely are present and possibly doing some damage in ME/CFS – she turned to those.
Three of the five, remarkably, were associated with one part of the immune system – the interleukins (IL-1, IL-12B, IL-4R). Another affects cellular energy production, and the last affects nitric oxide production – which is important in blood vessel functioning (vasodilation) and inflammation. The fact that all five – impacting energy, the immune system and possibly blood vessel functioning – fit this disease well was definitely encouraging.
Rare or Unique Variants
It got better, though, when the group searched for rare or unique gene mutations found in the ME/CFS group. Each person had an average of 14 rare or unique gene mutations. The good news is that the rare mutations, while quite variable, also made sense – hitting metabolic, immune, ion and mitochondrial pathways.
In keeping with the results of past studies, no rare mitochondrial gene variants were found. As we’ll see, though, it’s not necessary to take a two-by-four to the mitochondria to whack someone’s energy production – there are plenty of subtler ways to do that.

Detailed patient stories are providing clues that are assisting in the genetic analysis.
The fact that most of the rare gene mutations were unique to each patient presented problems. Determining which ones might be causing ME/CFS required an unusual kind of digging. It was time for Birch to get personal.
After taking notes on 60 of the 120 stories of ME/CFS presented on the Solve ME/CFS Initiative’s website, she realized that ME/CFS was more heterogeneous than she’d known. A variety of potential subsets jumped out at her – each of which could have a different molecular basis.
- About 1/3rd described an infectious type onset.
- About 10% never felt normal, but the problem didn’t get bad until they hit their teen to adult years. This group slowly got worse over time.
- Another small group described an extremely rapid and massive onset triggered by a non-infectious event – surgery, trauma or other very stressful event.
- A few people described cognitive problems so severe that they sounded like they had something like Parkinson’s.
- Another group described really severe pain.
- Another group had really severe orthostatic intolerance.
She needed some more information, though, and asked each person an additional 32 questions (!) that would help the researchers guide their analysis. She’s basically doing a precision genetic analysis – examining each person’s story in detail – and using that information to better understand their genetic results. Included were some open-ended questions which allowed the participants to just tell it like it is in their own rich detail.
She’s now analyzing that data, but even without it the preliminary genetic analysis provided some intriguing possibilities. Every one of those ten patients had genetic issues in three pretty darn exciting categories.
The Webinar
Energy Metabolism
The first was energy metabolism, a subject that’s getting more and more attention all the time. Another nice pattern formed when she found that three of the patients had a potentially very damaging and rare gene mutation that affected the AMPK energy sensing pathway – which studies have suggested may be compromised in both ME/CFS and fibromyalgia.
None of the genes have been associated with a disease before – which, in my book, counts as a plus. Intriguingly, these genes are closely related – one tells AMPK to ramp up, another to ramp down, and another has an intermediate role. That close relationship suggests that different kinds of damage to AMPK could result in the same kind of fatiguing problems.
AMPK ensures that proper ATP levels are present in our cells. As ATP declines during exercise, for instance, AMPK ensures that more ATP is produced.
Way back in 2003, Dr. Grahame Hardie (“Management of cellular energy by AMPK”) suggested that AMPK problems were present in ME/CFS at a MERUK Workshop talk. AMPK should get activated in our muscle cells during exercise, but more recently, when Julia Newton in the U.K. whacked the muscle cells from ME/CFS patients with exercise, AMPK didn’t respond…
In type II diabetes, AMPK activation issues produce “metabolically inflexible” muscles that have trouble switching between glucose and fatty acid metabolism. Researchers refer to the reduced skeletal muscle mitochondrial capacity that results as “mitochondrial overload”.
Interestingly, if AMPK is a problem, a wide variety of treatments may be able to help (see blog above).
Iron Metabolism
They found rare and damaging mutations in two people with ME/CFS in iron-metabolizing genes that could cause trouble transferring iron from the liver or in iron recovery. Because iron carries oxygen to our cells – which then use the oxygen to produce energy – it’s no surprise that iron deficiency diseases such as anemia can cause enormous fatigue.
If an anemia is present in ME/CFS, though, it’s a very different kind of anemia than what we’re used to. Birch brought up the interesting possibility that if anemia is present in ME/CFS – it’s not present in the blood – it’s present in the tissues.
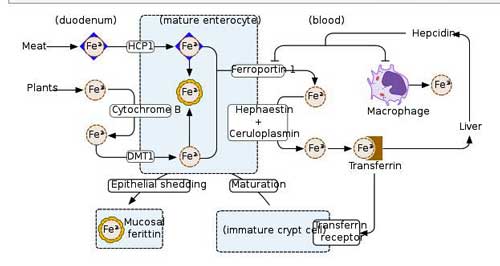
More rare mutations in genes involved in iron metabolism – another potentially energy-affecting pathway.
That was an interesting possibility. Throw in another admittedly very preliminary (and not genetic) report from an ongoing study at Ian Lipkin’s research center – and the iron issue gets even more interesting. Instead of examining the makeup of the genes we are born with, Lipkin is examining which genes are active.
In a possible tie-in, Lipkin found that several genes associated with iron metabolism were less active in ME/CFS patients. The differences were modest, but because they showed up in one pathway (that pattern thing again), Lipkin thought the ultimate impact could be significant.
In fact, Lipkin stated that, based on their “very early” data, they could predict that lesions in four parts of the iron intake pathway into the cell could be present. Not getting iron (and oxygen) into the cell could, of course, put quite a damper on energy production and wreak havoc with the resulting oxidative stress. Problems with that pathway could spell trouble in two potentially very important aspects of ME/CFS: oxidative phosphorylation (ATP production) and oxidative stress.
Glycogen Storage Disease?
So far, Birch had found rare and damaging mutations that could very well impact energy production in five out of ten patients in that study. Would the trend continue? Would we be so lucky? Rather remarkably, we were. In fact, Birch actually saved the best for last. In what she described as another surprise, mutations in glycogen storage genes showed up in two people.
Both mutations are very, very rarely found in the general population. One gene codes for an enzyme called enolase 3, which is associated with a glycogen storage disease (GSD 13) which, interestingly enough, shows up in adults (check), is characterized by severe muscle pain post exercise (PEM; check), and is associated with an intolerance of exercise (double check!). It’s one of two GSD’s associated with “exercise intolerance”.
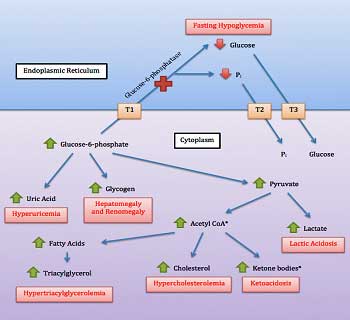
Problems with glycogen storage is another way to affect energy. Ron Tompkins will be assessing glycogen storage in ME/CFS muscle biopsies.
Birch was very calm, but I was about jumping out of my seat when she described a genetic disease with adult onset, severe muscle pain after exercise, and exercise intolerance.
The plot thickened further when she reported that GSD 13 is thought to be very (VERY) rare – as in present in only two families thus far. She noted that that fact is almost irrelevant, because, if no one is testing for this disease (it requires a muscle biopsy) – and few doctors probably are – it’s going to be rare no matter how many people are affected. It’s not uncommon at all to later find that “rare diseases” aren’t so rare after all.
If the mutation causes severe muscle pain after exercise and exercise intolerance, one wonders how many people with FM or ME/CFS may have been misdiagnosed? Will GSD 13 be like craniocervical instability – a rare condition or disease with a difficult diagnosis – which doctors just haven’t been testing for?
Of all the GSD’s, GSD 13 is clearly potentially the most relevant for ME/CFS. Most of the other GSD’s produce in young children low blood glucose levels, enlarged livers, swollen bellies, heat intolerance, slowed growth and muscle cramping/pain after exercise.
GSD 13 – which is not even mentioned in some medical websites, including the Association for Glycogen Storage Diseases (lol) and the Cleveland Clinic (but is in Wikipedia – go figure) – does not cause hypoglycemia, enlarged livers, swollen bellies, etc., but is one of only two GSD’s that causes “exercise intolerance” and “an increasing intensity of muscle pain over the decades” (check!).
It “appears” to be extremely rare. The website on which the most GSD 13 information is found states under epidemiology – 3 patients (!). The clinical pool is obviously very small, but the website states that muscle strength is usually normal, episodic elevations of serum creatinine kinase can occur, which are reduced and can be normal with rest, and that episodic rhabdomyolysis may occur. (Rhabdomyolysis is a serious event which occurs when muscle fibers rapidly break down and release so much muscle detritus into the blood that kidney failure can result.) Beyond that, it appears that we know little about the disease.
Lastly, another patient had a mutation in another gene associated with a glycogen enzyme (glucosidase alpha neutral C) that could impact glycogen storage, but not much is known about that enzyme or gene.
I don’t know if I particularly want the answer to be a rare genetic disease (lol) but I certainly want answers. Whether or not GSD 13 is present in ME/CFS, the potentialglycogen storage issue, as well as the AMPK and iron issues appear to open up more possibilities.
Conclusion
It’s important to note that Birch is not hunting for rare AMPK or ion metabolism or glycogen metabolism gene mutations – they are simply the ones that are popping up. They popped up in 7 of the 10 patients.
So far, ME/CFS is looking more like a glycogen storage type disease than anything else: rare, different gene mutations have popped up that could produce problems with energy production in different ways.

As Birch suspected, rare gene mutations that could affect energy production were found. More work needs to be done to validate the findings.
Of course, we don’t know if these gene mutations are having an effect or if they’re present in other ME/CFS patients. The body is complex with many redundancies. Just because you have a rare and potentially damaging gene mutation doesn’t mean it’s impacting your health, and, indeed, Dr. Birch warned that the study is quite small and that as more patients are added and they dig deeper molecularly, some of these avenues may not pan out.
Dr. Birch emphasized their desire to collaborate with others and stated they were actively talking to other ME/CFS researchers as well as geneticists outside the ME/CFS world – and then mentioned two. She said she spoke to Nancy Klimas at the last NIH conference and to a UK researcher involved in a very large genetic study.
That’s just the tip of the iceberg in this evolving field. Avindra Nath at the NIH should certainly know about this research – he’s doing muscle biopsies – possibly right now – and genetics work; Ron Tompkins of the Open Medicine Foundation’s Collaborative ME/CFS Research Center at Harvard will be doing the deepest dig in ME/CFS patient’s muscles yet; Ian Lipkin would probably love to hear of more evidence of iron metabolism issues; Allan Light in Utah has been involved in several genetic studies and a family study is underway at the Bateman Horne Center. Of course, there’s Ron Davis at Stanford – our in-house genetics expert – who is overseeing a personalizied approach to the genome in studies of families with ME/CFS, as well as other genetic studies with the Open Medicine Foundation’s help.
When contacted, Ron Tompkins said his team would be assessing glycogen issues in the muscle biopsies they’re doing.
The group’s findings were presented at a clinical genetics conference recently and were “received really well”. The presenter said the frustrated doctors (they are frustrated, too) were quite eager to get some potential answers for this disease.
Pilot Studies Work!
The Ramsay awards are designed to open up new areas of inquiry and provide researchers with the ability to gather the data needed to get major NIH grants. No data – no big grants! Grant packages like the Ramsay Awards are small, but potentially very powerful. They’ve historically played an important role in our continuing efforts to understand ME/CFS. Jarred Younger is just one researcher who got his feet wet in ME/CFS using pilot study grants. (He recently received another large NIH grant.)
This study is another win for the SMCI’s Ramsay Awards and for the SMCI’s newest research director, Sadie Whittaker. In her first round of Ramsay Awards, she chose well with this one.
Keep the Information Flowing!
Support This Website!


Hi Cort – thanks once again for an eye-opening article. I’ll read it a few times to really get all the content you sent. I did immediately, though, flag one comment that confused me. You wrote that (and I’m referring to it, not quoting it) muscle weakness is not an issue in ME/CFS. That confuses me first, because under the old definitions of ME/CFS, “unexplained muscle weakness” was one of the list of symptoms; and secondly, because I do find my own muscles suddenly became weak (and it hasn’t seemed to be possible, despite frequent attempts at PT and exercise etc, to get them stronger). Have they changed the list of symptoms now for ME/CFS to not include “unexplained muscle weakness”? Thanks.
My understanding is that studies have generally found that muscle strength is OK. Muscle endurance may not be good but quick tests of muscle strength have found it to be relatively normal. It’s been awhile since I’ve read anything on that, though.
In my case, as long as my disease resembled somewhat the disease described by the term “Chronic Fatigue”, muscle strength was not affected. Only endurance was.
When the disease went for the worst, extreme muscle weakness was the new normal. It may be just a question on what subgroup studies affected: the moderate or severely ill?
Muscle strength less in this study – https://www.ncbi.nlm.nih.gov/pubmed/21166613
And this one:
https://www.ncbi.nlm.nih.gov/pubmed/10945803
Despite my clear memory that muscle strength was retained I can’t find any evidence that it is!
Cort,
My muscle strength really was a major symptom, and its never gotten back to normal…I think it is a symptom for many of the most ill patients..I would include it.
Re muscle weakness, thanks for the studies you included. They go back several years, and seem to include some of the old assumptions about CFS patients becoming weak because they didn’t do anything, rather than not doing anything because they had suddenly become weak – which was certainly the case in my history. One of the aspects of David Systrom’s research and his suggestion of Mestinon as a possible treatment is that the drug is meant, bottom line, to make the muscles act stronger. Fingers crossed.
With me, I’m having progressively less muscle strength. (It’s not from not moving as I walk a good bit.) Both my sis and I test positive for a rare genetic gene that causes ataxia which includes muscle weakness (found in one Amish family according to research – and we aren’t Amish) to point, with me, of inability to walk at times. My legs give up picking up. I can shuffle feet, but not pick up leg. Can’t get into a car when this happens and have to physically pick up legs with arms or have someone pick up what seems to be “dead” legs. It’s like I used up the energy and there is nothing left there. This also is connected to mitrochondrial dysfunction (energy in the cells). Along with leg weakness, there is neuropathy. This runs in my family. My dad and his dad both had it from their feet up past the knee and fingers past elbow. Both me and my sis seem to be having the same thing with neuropathy but my sis doesn’t seem to have the leg weakness.
Issie
Uh, Cort. The reason people can’t walk, or stand, is because the pain gets so bad because they’re glycogen stores are depleted from NOT RESTORING THEIR LEVELS.
Like carb-loading?
And duh, as others have said, muscle WEAKNESS eventually is obvious. Tell Whitney Dafoe and others that their muscles aren’t weak.
Sheesh.
Hi Cort,
I am so grateful for all the work you do. I am amazed at how well you translate all this complicated information. Keep up the good work!
Deane
Thanks Deane,
I’m very lucky to be able to be able to do this. Life would be more difficult without it…I really am very lucky to have something I can do with this disease. Hopefully I will be able to keep it up.
Neil McGregor also mentioned iron metabolism in his recent Emerge Australia presentation.
Cleveland Clinic uses uncooked Corn Starch in GSD 13 the same product is used in Hereditary Alpha Tryptasemia (HFI) a Genetic Born illness passed on from parents…If the same type of GSD 13 they can lead Normal productive lives, if HFI all along Cancer
of the Liver is likely & must be treated to survive I know EDS has HFI I have seen countless with this now but rarely tested for either…There are HFI Groups on Facebook I do not like Fruits never have also Tomato is a fruit it is also in countless
products even vitamins minerals there are places in Germany online to purchase safe vitamins…It may be GSD13 combined with HFI
typo I meant to write (HFI) Hereditary Fructose Intolerance instead they use Corn Starch as a treatment
I am most interested in how this might relate to mast cell activation syndrome. I am 80 years of age and was diagnosed with MCAS the first of the year. Looking back I am sure I have had this illness all my life. As a young person I was anemic and had B 12 shots some times up to five a week. I struggled with different symptoms off and on but managed to work, have a family etc. Later in life I was diagnosed with Lyme disease and went through some horrific treatments and were told that I would never be troubled with it again. Several years after that, I had two separate surgeries for hiatal hernia and in one they thought I would not pull through and did not have a clue as to why. After pumping me full of three pints of blood, I was ready to dance. In the summer of 2018, l begun to feel extremely tired with what felt like the flu 24/7. Made an appointment for a check up which included blood work EKG, eeg and stress test all pass with flying colors. Why do I feel so bad was my next question and because of my age I got the usual reply…..well you are 79.
I thought it over for a few days…did some research and talked with a cousin that had just been diagnosed with Hats and made an appointment with a allergist/immuneolgist. That started a long series of test included bone marrow biopsy. Some of the test were negative but the tryptase which was staying in the mid + twenties along with the itching and the ilgs, etc. I am now on numerous meds along with xolair shots.To finish this, I can see similar behaviour between your cfs and mcas and wondering how i can go about having this gene testing. For so many years I have been treated for so many illness and now of course it has always been MCAS. Can’t tell you how many different doctors would say you have cfs and I would said no I don’t think so. But since the similarities are there I would like an answer about the genes.
A quick note…I am from Mobile, Ala and have lived in the Tampa Bay, Fl. area for years…and my mother was a Ramsay.
Thanks for listening to my long winded story.
Carol Grice-Curran
I’m from Mobile too. My mom had MCAS and so do I. I think alot of the problem is the toxins we were exposed to there with Paper Mills, mosquito poison spraying and who knows what else. I also feel partially some vaccine I had as a child triggered epigenetic response that never got better. There is generations of issues in my family and I think alot may have been environment related.
BTW Carol, I love your paintings! So much how things used to be with the seascapes.
I’m back in South now. Humidity seems to make everything worse. But not having as much issue with dehydration as I did in the West. Wonder why…. 🙂
Hoping to get with these genetic docs as they are only an hour from me in Birmingham.
Issie
So, if this is true, why now, and in recent decades are these conditions presenting? Why didnt grandparents and parents have “CFS”, if they had the same genes? I’m not arguing against it. I have GBA, HFE, A1AD, GSTM, SOD, polymorphisms. But why am I and so many others ill, when our parents, and grandparents were not, if this is related?
These are my off the cuff not terribly informed guess.
The gene mutations may only apply to some patients. If that GSD 13 disease is present it may only be present in a small percentage of patients. (Even if it’s a small percentage, though, it could be helpful for the rest of us as removing those patients will result in cleaner studies.)
How damaging mutations in a gene are depends, I believe, on other factors as well. A damaging mutation in one person may have no effect because something else in the body is making up for it or causing it not to fully express itself. In another person an epigenetics change could cause that mutation to more fully express itself. It’s possible that epigenetic changes effect other genes in the same pathway causing the original mutation to have more profound effects.
Plus, while many of us don’t have this disease in our families enough people do have it in their families for Dr. Birch to think that genetics does play a role. Genetics may, therefore, play more of a role in one group than others.
We also saw that a mutation in one gene can have different effects in different people. It’s possible that your ancestors had this mutation but it showed up differently in them…
One issue is that many genetic conditions, can require two copies of the same gene, from either one parent, or from each side combined. Some conditions only require one copy from any side of the family tree. So for example, I have a copy of the GSTM Polymorphism, from both family lines. Meaning I am +/+, whereas my parents may both be +/-. They are carriers of the same thing. There is argument that one copy may create a milder version of the same thing. That certainly applies to A1AD and seems to be suggested in HFE.
These sorts of paper interest me, because they show perhaps what is I guess is coming next. Where rare conditions are identified more often, and also the question of how they combine is raised to:-
A1AD +HFE
https://www.ncbi.nlm.nih.gov/pubmed/3485129
Gaucher + HFE as rare sub-group of two rare conditions.
https://www.sciencedirect.com/science/article/abs/pii/S1079979616302066
Gaucher + A1AD
http://www.jgld.ro/wp/archive/y2019/n1/a19/
So its interesting to think about how this all goes together. But also how they influence more standard tests designed to identify one of them.
What begins to puzzle me though, is how things like HHV6, EBV or Lyme will fit with these rare conditions and combinations too? Is this suggesting they are not involved in those cases, or explaining why they are?
“What begins to puzzle me though, is how things like HHV6, EBV or Lyme will fit with these rare conditions and combinations too?”
There seems to be an interconnection between the immune system and oxidative phosphorylation.
That is not that unlikely. A strong immune response requires the white blood cells to produce plenty of oxidative stress to kill or destroy the suspected pathogen. Doing so requires plenty of NADPH. Cleaning up the collateral damage this oxidative stress creates requires plenty of glutathione. Recycling oxidized glutathione to its active form again requires plenty of NADPH too.
So a strong immune response requires the energy metabolism to decrease NADH production in favor of NADPH production. One way of doing so is to decrease oxidative phosphorylation (aerobic mitochondrial pathway) and turn up the pentose phosphate pathway.
Many of the genes mentioned in the study affect either the immune system, energy metabolism or both. And
HHV6, EBV or Lyme are known to produce stuff the immune system can react too on a chronic basis even if the infections are latent.
“How damaging mutations in a gene are depends, I believe, on other factors as well. A damaging mutation in one person may have no effect because something else in the body is making up for it or causing it not to fully express itself. In another person an epigenetics change could cause that mutation to more fully express itself. It’s possible that epigenetic changes effect other genes in the same pathway causing the original mutation to have more profound effects.”
This is how it was explained to me. Dr. LIGHT in Utah is doing genetic studies on families. My sis and I have 50% same genes. I may have 2 copies of one mutation and she only one. But either of us may or may not express symptoms because other genes may be taking over that function. They can measure the function and if it is expressing dysfunction or not. Of all CFS patients tested, I was in lowest function of 20% of all tested. I had many mutations and they were expressing. My sis were not.
We both had a very rare mutation that had only been found in one Amish family. Both of us showing it actively affecting us. It causes mitrochondria dysfunction and ataxia. Could be contributing to our neuropathy and pain.
I would love to contact the doc looking in to these rare gene mutations. If we have one, we may have more.
Issie
Whoa! How could I forget Dr. Light and the genetic studies going on in Utah? Thanks for the reminder – I just put them in the blog.
Darren,
I would suggest the possibility that some dietary factors may be involved. Our parents and grandparents didn’t eat foods high in omega six fats or synthetic fats like those in margarine — huge in the late 1960’s and on — (which mess w/mitochondrial function big time) and inhibit or lower glutathione, SOD,etc. They also didn’t eat or drink large amounts of high-fructose corn syrup, but used sugar instead. They weren’t iron-phobic like so many are today. Note how many multivitamins or mineral supplements don’t contain iron — something needed for the immune system and the krebs cycle. On and on and on…
They didn’t have cell phone towers and wi-fi, etc., etc. And lastly, stress levels were lower in general. One parent worked, while the other stayed home. All that’s changed…
Yes thank you.When it comes to glycogen storage and lipid storage disorders, it is certainly interesting to wonder what dietary changes and additives have done to those conditions over the years. Interestingly many of them are to do with the liver.
I have no idea if this applies but when you say liver I remember that the liver where Ron Davis thinks whatever in the blood that appears to be affecting our cells may very well come from.
Also the corn syrup we eat today has been grown with glyphosate herbicides, unless it is organic.
It is creepy how the use of glyphosate herbicides on food crops coincides with the rise of ME/CFS and autism.
they were likely carriers & passed both bad genes on & in some it skips even generations or Family members like EDS does
My daughter is 23 and has a classic case. Healthy until 18- contracted EBV in college, had a traumatic physical event and of course- consumes a diet chock full of sodas, energy drinks and inorganic foods. Her health and life quality has plummeted as all the usual suspects and symptoms are present. Too many u successful “specialist” visits prove inconclusive and have her a non- participant due to the emotional toll of the letdowns. What’s a loving Dad and Mom to do? Where do we go from here?
Scared –
Dad
That diet is certainly not helping. Some people occasionally have huge gains via diet…Check diet section on the Health Rising’s Treatment Resources – https://www.healthrising.org/forums/resources/categories/diet.172/ – in particular ketogenic diets and The Wired to Eat book…..Sugar increases inflammation and can cause depression! Good luck!
As Kelly already mentioned, the amount of fructose in a “modern” diet is hugely higher in the last decades compared to only 1 or 2 generations ago. And that’s just one dietary factor.
When it comes to the immune related genes, the same question can be asked regarding to allergies: “why are allergies soooo much more prevalent nowadays (especially among youngsters) compared to only 1 or 2 generations ago?
Allergies carry a very large immune component as well. And their diagnosis very often is confirmed by blood tests so it’s not only imagination or fashion. Something seems to turn up our immune systems, and with “our” I mean the majority of the Western population.
“Three of the five, remarkably, were associated with one part of the immune system – the interleukins (IL-1, IL-12B, IL-4R).”
I admit I did search specific for it so there could be plenty of bias here. But looking up “IL-1 IL-12B IL-4R neutrophils”
A few interesting finds:
https://www.sciencedirect.com/science/article/pii/S0091674919302039
with title “IL-4 receptor engagement in human neutrophils impairs their migration and extracellular trap formation”
“IL-4R stimulation on human neutrophils by IL-4 or IL-13 decreased NET formation. • Stimulation of IL-4R on human neutrophils downregulated their CXCR1 and CXCR2 expression and impaired their chemotaxis to CXCL8 in vitro”
https://www.hindawi.com/journals/mi/2009/193970/
“Interleukin (IL)-4 is a cytokine known mainly for its anti-inflammatory activity. Using the in vivo murine air pouch model, we found that IL-4 significantly increased the number of leukocytes after 9 hours of treatment, consisting mainly of neutrophil (60%) and monocytic (40%) cell populations.”
https://www.researchgate.net/publication/296911462_Interleukin-4_prevents_apoptosis_of_human_neutrophils
“Interleukin-4 prevents apoptosis of human neutrophils”
Now messing with neutrophils and NETosis is messing a lot with innate immunity. Note: wether something increases or decreases in above links does not matter much: the gene deviation doesn’t state which way it goes.
Also remarkable:
https://www.sciencedirect.com/topics/neuroscience/interleukin-4
“In vitro, IL-4 increases type I collagen production by human fibroblasts”
-> A link to the EDS community?
“They demonstrated in a mouse model that IL-4 is a crucial factor in muscle growth.”
-> Weak link to combined ME/FM patients?
“Interleukin-4 (IL-4) … …to the regulation of the adhesive properties of endothelial cells via VCAM-1 and the regulation of ion secretion by intestinal epithelial cells.
-> Now that would be a significant link to changed gut immunity and response to gut bacteria IMO
“Treatment of intestinal epithelial cells T-84 with IL-4 (Wisner et al., 2008) or IL-13 increases the expression of claudin-2 and paracellular permeability through a process mediated by the PI3K pathway”
-> Increased intestinal epithelial cells paracellular permeability sounds a bit like an element of leaky gut syndrome.
“Ligand binding induces heterodimerization of IL-4R, leading to the activation of cytoplasmic tyrosine kinases (Janus kinase family, Jak), which in turn induces the phosphorylation of cellular substrates”
-> Now this gene also seems to mess with phosphorylation aka energy production
“Indeed, quiescent microglia, which reside around the proliferating NPCs, may become activated by IL-4”
-> IL-4 is involved in neuronal regeneration too
One wouldn’t want these IL-4 genes go wrong. On a side note IL-12 seems to be associated with Bechet disease (plenty of aphtuous ulcers and more).
Cort, thanks for the good overview of Dr. Worthey’s and Dr. Birch’s research. I watched the video and indeed it is worth viewing–if only to give people an idea of how geneticists sort through millions of data points to find correlations to certain disorders.
I copied down the list featured on one ‘slide’ and went to my sequencing to check if I had any of the mutations she had found. Nope. None. I did however, check Dr. Phair’s IDO2 variants in his ‘metabolic trap’ theory and I had two high impact mutations–just like he predicted.
Before everybody gets all excited that they are hot on the trail of identifying the problematic gene, I think it quite likely, especially because of the varying presentations of ME/CFS, that the disorder will be found to be polygenetic–at least in the majority of cases.
Chasing ultra rare variants may or may not be the answer, as fatigue could be the result of several (or many) variants–and not necessarily rare. Everybody has variants, some rare, and most not–and some have many–but are not much affected as might be predicted.
Honestly, of the billions of genes, only a relatively small handful of single variants, comparatively, can be directly linked to diseases–and then again, not always.
Genetics is such a new and complex field, and all the afore mentioned is probably why, when I pulled out a stack of my own suspicious mutations to show my new Stanford doctor, he tossed them aside and said something to the effect of ‘not enough information.’
My position is, ‘So what? You take what you can get and go from there! Because you don’t understand doesn’t mean you stop looking!’
🙂
I agree – I think most people do think the genetic contribution to ME/CFS is going to be complex, involving a variety of genes. Plus there’s the epigenetic and gene expression component – it’s incredibly complex which is why, I imagine, organizations focused on this problem like Hudson Alpha exist.
Here’s the bio of Jim Hudson from the Alpha Hudson website:
Equal parts scientist and businessman, Jim Hudson’s investments have shaped the advancement of biotechnology around the world—but especially in the state of Alabama. Hudson founded and served as chief executive of Research Genetics, Inc., which in the 1990s became the world’s leader in genetic linkage products and an integral partner in the Human Genome Project, the international effort coordinated by the U.S. Department of Energy and the National Institutes of Health to identify the sequence of the DNA found inside human cells. Following Research Genetics’ 1999 merger with Invitrogen Corp. (now Life Technologies, Inc.), Hudson remained committed to local entrepreneurs, serving as a mentor and advisor to companies in industries ranging from genetics research to Internet services.
https://vimeo.com/178471898
Nancy B, which sequencing test did you do & what test or home test kit would pick up the GSD type Gene mutations?
Hi Cort, Interesting pilot study! Thanks for the summary. I’m wondering what tissue did they do the genetic testing from i.e. blood, muscle? The reason I ask this is, as a for instance, the TFRC gene is expressed differently in different tissues, it has different purposes based on which tissue it’s in. Their up and coming messenger RNA and protein expression study will be the most important piece of this. (You can have a mutation and it means absolutely nothing.) Understanding a gene mutation requires knowing how it’s being expressed in the body, if at all, and whether ie it results in gain or loss of function. Again, as an example, the TFRC is also implicated in hemachromatosis given how it is expressed and where, this is an iron overload syndrome, not an anemia. (And, is an important disease to rule out having an insidious onset, and can be diagnosed As ME/CFS at the outset …. ) Just to give you an idea of how complicated going from gene mutation to understanding expression and phenotype can be take a look at this website: https://www.uniprot.org/uniprot/P02786
Interesting! I imagine that it was the blood but don’t know for sure. I do remember that most of the mutations highlight were deemed, I can’t remember exactly how she termed it, but something functionally dangerous which I guess means they had the ability to damage the functioning of the protein they were associated with.
When you put everything together it is amazingly complicated!
I would like to see what response comes back from Ron Davis especially with his Team looking at this now in muscle biopsies I am sure he will say
if it is a huge find or not…Do you think Cort from the 10 people it looks possible that we all have GSD, I find it odd though Corn Starch & Glucose
iv is used in HFI Hereditary Fructose Intolerance…It could may be both involved
Interesting. I would be totally shocked if we all had GSD but it certainly seems possible that some of us do or have some problem with glycogen storage. What really intrigued me about these findings – and we should be clear that they are preliminary and may not pan out as more work is done – are the number of different ways to affect energy. If I had to bet I would be that there’s end state – ME/CFS/FM and there are a number of ways to reach it. –
Here’s a good reference for the *8* types of GSD:
http://www.chp.edu/our-services/transplant/liver/education/liver-disease-states/glycogen-storage-diseases
At 3:50 in the video she says “When you take a blood sample and do the sequencing…” so probably they used blood but she goes on to say they are experts in the analysis of the genetic data and not the sample collection and sequencing.
James, they first do muscle biopsy then if Positive move onto the panel of GSD blood sequencing DNA so both are used in diagnosis also I read an EMG in Neurology is done as well it looks for a specific thing not sure now what it is though
And to think the Victorians thought there was nothing interesting left to be done in science.
Thanks for the laugh
🙂
Dear Cort. Thanks again for making pretty compicated stuff availeble ?
A simple question from a simple foggy mind;
if genes are involved: Are there any hope for cure or sympthomsrelief?
Could bonemarrow-transplant hypothetically work? Has anyone ever tried that?
Or are we then “doomed”? ???
Thank You again for Your great work ?
I don’t think we’re doomed at all. Nancy Klimas is testing two drugs, there’s the Cortene drug, in another SMCI funded study, Vince Lombardi is looking for evidence that current immune drugs could, FLuge and Mella are continuing their drug trial and Ron Davis is testing drugs with the nanoneedle at Stanford. If AMPK is involved lots of possibilities may be present plus Jarred Younger is checking out an even better form of LDN.
There are lots of possibilities 🙂
Hang in there!
ThanksCort. Any idea if carnitine deficiency would fit in here? I know it is sign of FOD (fatty oxidation disorders) but wondering if it can also be finding in glycogen storage. I was too fragile to undergo muscle biopsy at time the serum level of carnitine was found to be low. And my profound response to prescription Carnitor was enough that my neurologist said no need to muscle biopsy for time being.
I too have had successful use of corn starch to manage overnight hypoglycemia.
Since I am hEDS, the GeneDX WES that was done was with a focus on Ehlers-Danlos and related connective tissue disorders. Within that scope, FLG (filaggrin) defect and ENG (endoglin) defect were found.
The carnitine connection is really puzzling me. I know Jennifer Brea mentioned in a recent blog post she too is on Carnitor.
“I know it is sign of FOD (fatty oxidation disorders) but wondering if it can also be finding in glycogen storage.”
I’ll give it a try:
Basic rest metabolism is mainly using fat and glucose as fuel. Glucose is used in an aerobic way.
Increasing activity increases the use of glucose in both an aerobic and anaerobic way and the use of amino acids as fuel to supplement on top of this basic metabolism. Anaerobic glucose consumption is an expensive process producing increased waste. I also strongly believe very fast spike-like use of amino acids for fuel is quite a dirty process producing plenty of waste.
Now both processes that impair glucose delivery or oxidation and processes that impair fatty acid oxidation are going to result in turning to spike-like amino acid consumption a lot more even when doing small activities, with all potential waste and problems associated with it.
Thanks Cort, this may explain why ketosis helps some of us . ???
I don’t know where – but I imagine it’s in there somewhere. The problems some people such as myself have with carbohydrates must be a huge clue… to something 🙂
Carbs destroy me especially Pasta Bread Pizza, if I have bouts of Insomnia I’m Cured with Carbs they make me so profoundly tires I wonder if it is the
Sugar contents I know I do not have Lactose issues Negative & Negative to Celiac but only issues without Carbs is constipation…
In reply to Darren M. ME/CFS may have been hidden in the diagnosis of Neurasthenia. My great uncle had it. The army took it seriously enough that he and other men with the condition were exempted from serving in World War one.
Wow…the first person I’ve heard of with a relative with neurasthenia! It definitely runs in your family. It would be great to get researchers your genetic data.
The only relative with something similar I had is a distant cousin of my mothers who decades ago had severe chemical sensitivities. She must have had a rough life…
Cort, thank you for the vimeo link to AlphaHudson. After watching the first video, one can go down the list on the right hand side of the page and get a sequence of very short genetic tutorials. Lots of great information packed into them–almost too much! They are a good way to see just how complex genetics can be–and these are just snippets!
I’m sure you know about the NIH’s All of Us program to sequence one million individuals for a research data bank. I’m not sure what it takes to get access to this information, but hopefully it isn’t so restricted so it can be useful for (often small budget) researchers like the ones trying to figure out ME/CFS.
If anybody want to study genetics, I believe Khan Academy has free on-line courses…The more that gets known, the closer we will be to personalized medicine!
Thanks for the tip Nancy, I didn’t know about those and will check them out. 🙂
Odd thing though the Gulf War Illness Vets in the UK Diagnosed in Endocrinology with Glucagon Stimulation Testing of the Pituitary they use Hormone injections daily
Growth Hormone &Testosterone. The Endocrinologist involved went to Dubai to continue his work when dismissed in the UK…I do not think they are well though
I have had cfs since at least 1989 with high Ebv tigers then. I did however already have symptoms in my 20’s. I am now 71 and have a 34 year old son who I think may also have immune system issues. Shingles at age 6, ibs in his twenties and interesting a very bad case of Rhabdomyolysis at age 30. He was hospitalized for 8 days until his numbers were safe but not normal. No expert could determine why this happened. He lightly lifted weights two days in a row for 20 minutes to de stress during a very stressful job period with little sleep. According to experts this should not have caused the Rhabdo.
There you go – a possible familial connection – not ME/CFS – put a strange incident with his muscle metabolism…Very interesting!
I hope you will warn your son to never, ever have statins! They can cause Rhabdo.
Your conclusion that “ME/CFS is looking more like a glycogen storage type disease” doesn’t seem borne out by the rest of this article.
Does that correspond to other work you’ve covered, or is it more of a personal relevance to you, with symptoms of exercise intolerance and muscle pain?
From the above, the AMPK aspects seem to have more general relevance and incidence. And personally, the tissue iron deficit anaemia is more interesting – sounds like it might be part of my picture, perhaps fitting my slightly odd aerobic response, throughout my life. Are the Lipkin details you mention all covered in your recent article here?: https://www.healthrising.org/blog/2019/04/15/nih-chronic-fatigue-lipkin-davis-prusty-oh/
Also reassuring to hear that as many as 10% of pwME/CFS were gradual onset, like me; post infectious only 1/3 (given that I excluded CFS for a long time because I assumed infection was generally necessary).
But we should take those percentages with a big pinch of salt, right? I mean, I don’t know how the SMCI stories were selected, but they’ll all be long term suffers and likely a biased towards showing a diversity of patient phenotypes. Right?
That was a bit confusing I admit. I didn’t mean that ME/CFS was a glycogen storage disease but that it was looking like that type of disease in which a bunch of entirely different genes were contributing to different flavors of ME/CFS.
I agree about AMPK – since we have some past studies that suggest it’s involved, it would be my top pick, for what’s it worth, to be the issue that affects the most of us.
Yes, the Lipkin details are in that post.
Never exclude ME/CFS because you have gradual onset! Although infectious onset is most often mentioned, gradual onset is not uncommon at all. My ME/CFS onset was gradual over a month or so and was associated with an infectious trigger or any trigger that I can remember. I was doing quite well at the time in fact!
For sure, we’ll need a lot more stories. I hope to work on that 🙂
Oh I see, “looks like it’s similar” to, rather than “like it will turn out to be” (a glycogen storage disease). Cool.
And my “gradual” was decades, merging with delayed sleep phase difficulties (and very late dyslexia and ADHD-PI diagnoses).
Oh, did you have a hand in creating those stories (or you mean to post your own)? These?: http://homecfs.solvecfs.org/
“my “gradual” was decades”
Mine too, depending on where you start counting.
Looking back, some parts of this disease started showing up during childhood. But nobody would have recognized it as (part of) a disease back then.
For more then the last decade, it did fit the definition of CFS. For about four years now, CFS is to loose a name and symptom list to fit the disease. I can call it nothing but ME now. Luckily I’m hidding slowly back to the years before I could no longer identify with CFS.
I have ME and the type of exercise that takes me down is strenuous exercise.
I can pull small weeds, snorkel or leisurely swim for extended periods of time, but I can’t sprint in the pool, scrub a stubborn stain from a pan, tug or pull a large root out of a garden bed, lift or hold heavy things for long. It’s intensity, if that makes sense.
How can a rare gene mutation suddenly take down hundreds of people in a small ski resort town?
@Erik Johnson:
A bit of topic here, but on https://www.healthrising.org/blog/2019/05/29/spinal-stenosis-chronic-fatigue-fibromyalgia/#comment-874284 near the bottom of the comment section, as a comment on Esther Siebert I posted a comment that may be of interest to you.
Did you consider an unidentified and hence untreated outbreak of Legionnaires’ disease to be a possibility? The Lake Tahoe outbreak was in a period that this disease wasn’t that well known yet.
The disease ticks quite a lot of boxes with what you describe. The bacteria is established within amoebae in a symbiotic relationship. Both algae and mold belong to the class of amoebae.
The disease also has quite an overlap in symptoms with ME, making it possible that the early infectious trigger morphed later into post infectious ME without being obvious that there was an initial infection stage and a following ME stage.
There are lately few new reported ME cluster outbreaks IMO. That fits with the observation that Legionnaires’ disease is mainly spread with cooling installations, hotel and pool heating systems and likewise. An industrial cooling tower taking in algae overrun lake water would pose a big risk. At least over here there are stringent rules now for all installations at risk in order to reduce spreading Legionnaires’ disease. Also outbreaks are far more recognized and treated now, labeling it non-ME and reducing it’s spread and improving it’s treatment.
Ski resort town you said?
Changes are small, but artificial ski machines using lake water might be an effective spreader of Legionnaires’ disease too under certain circumstances.
For example:
* Lake water collected and stored during summer or fall, containing sufficient amounts of algae.
* Pipe inlets covered in algae.
* Lake water collected in a well with extensive permanent algae growth or contained by a dam covered with it and inlets close to the dam wall.
* If the lake is far lower then the ski resort then using straight lake water in spring might do the trick too in sunny south California.
* Using the snow machines during night and having artificial ventilation in the houses.
* Worse: using the snow machines during day in spring with well above freezing temperatures.
Those snow machines are very good at creating really tiny droplets of mist out of water. They are designed to do so. They are also designed and placed in a way that droplets and the snow formed out of it stay close near the surface where people breath them in.
Granted, artificial snow machines seem an unlikely source of Legionnaires’ disease (also known under other names), but when looking into it:
https://en.wikipedia.org/wiki/Snowmaking
“invented the snow cannon in 1950… …Snowmaking began to be used extensively in the early 1970s.”
If it also was used in your ski resort around that time, it may fit the timeline.
https://hydrochem.com.au/resource/how-can-ice-machines-spread-legionnaires-disease/
If ice making machines can spread the infection in hospitals, then snow machine in ski resorts may be less unlikely suspects then it would seem at first sight.
This source states the compressor heating the water was the likely source of infection on the ice machine’s case. It also states “Severely ill patients are often offered ice to suck on to rehydrate and moisten their mouths, which leads to aspiration (the breathing in of the water particles), and infection where those water particles are contaminated with Legionella.”
-> So the bacteria can survive some amount of freezing.
In the snow machines case, the bacterial source may already be at the lake or reservoir the water comes from.
Also, from https://en.wikipedia.org/wiki/Snowmaking the snow machine itself has two sources of heat that I did identify:
* “The next step in the snowmaking process is to add air using an air plant. This plant is often a building which contains electric or diesel industrial air compressors the size of a van or truck. However, in some instances air compression is provided using diesel-powered, portable trailer-mounted compressors which can be added to the system. Many fan-type snow guns have on-board electric air compressors”
-> That’ll be the worst offender
* “Snowmaking begins with a water supply such as a river or reservoir. Water is pushed up a pipeline on the mountain using very large electric pumps in a pump house.”
-> Less important source of heat IMO
Improved water treatment techniques (biocides) and improved snow making machines (more economic in power consumption = less waste heat and improving techniques to form more snow and loose less water as vapor) may have reduced the problem in later years a lot.
Good resource on the disease: https://www.who.int/news-room/fact-sheets/detail/legionellosis
The answer is really very simple. If you have a mutation which inhibits your abiilty to fight off a certain kind of pathogen, then if that pathogen sweeps through a town It will hammer people with that mutation.
Same applies if there was some toxic event and your genetic makeup made it difficult for you to detoxify that toxin or if there was a big fire and your genetic makeup predisposed you to respond to smoke with asthma.
Just curious, on the topic of the Incline Village outbreak, was there ever any data or study of that cohort who presented with or developed POTS (i.e. – only some, most, the majority, etc)?
Just curious since if I remember right in Osler’s Web Dr. Peterson/Cheney saw around 185 CFS/ME patients in that 1984-1985 timeframe (this is going off of memory, I’m probably off) and was always curious how many were orthostatically intolerant in that specific outbreak.
@ErikJohnson
(also posted below but realized this may be more applicable here)
Just curious, on the topic of the Incline Village outbreak, was there ever any data or study of that cohort who presented with or developed POTS (i.e. – only some, most, the majority, etc)?
Just curious since if I remember right in Osler’s Web Dr. Peterson/Cheney saw around 185 CFS/ME patients in that 1984-1985 timeframe (this is going off of memory, I’m probably off) and was always curious how many were orthostatically intolerant in that specific outbreak.
Both Dr. Cheney and Dr. Sarah Myhill [in the UK] found 90-93% of ME/CFS patients had PFO, patent foramen ovale.
If that was so then they were unwise not to pursue it.
Thanks for reply.
Although Dr. Cheney is deceased, Dr. Myhill currently lives in the UK, with website.
I requested that Dr Cheney follow up on this about 25 years ago, after Dr Fred Herman, FM specialist from Sacramento CA had a decompression sickness incident while scuba diving in relatively shallow water.
People with a PFO are particularly susceptible to ‘the bends”
But got no reply from his office.
Anything that shows up so strongly should be pursued imho.
As well as my great uncle having neurasthenia, my sister has fibromyalgia, like our grandmother (fibrositis). My sister’s daughter has ME/CfS. Our father and other sister had/has four autoimmune conditions between.To confuse the issue further this involves three different family lines.
Somebody really should study your family intensely! Family studies can tell us what gene or genes is responsible but more importantly what protein is involved – which can tell us so much about a disease.
The researchers need to find you and your family, seems to me. I wonder if their studies are still open.
Hi, I’m in the uk and have lots of muscle problems but more interestingly I carry multiple types of GSD (one of which is a homozygous pair of errors of unclear clinical significance). I have recently had my WGS done and am willing to send the files of all errors if this helps with more research and potentially gives me a answer
Wow…So interesting…They will definitely want to hear from you Pippa. I forgot to mention it but the team is moving to the University of Birmingham at Alabama and after they get set up there there will be asking for whole genome samples.
Pippa, does that mean now you do have a diagnosis of GSD & what type(s) are they I am now in the UK I would like to know more about your findings you had done I am in Southampton area
To Issie, back to muscle strength, and your addition of peripheral neuropathy into the mix. I “got” peripheral neuropathy with the onset of ME/CFS. It was rather creepy, actually: during one week, the numbness and tingling starting in my foot, day by day progressed up to my knee. Thank heavens it stopped there. The neuropathy in my arms just gradually showed up, not as dramatically. But the weakness is in my arms, not my legs. My hope is that mestinon will do as advertised. I’m taking it in liquid form, very very very slowly introducing it to my body. Still in first two weeks, still at one-quarter tsp per day. After that, twice per day. I’m guessing it will be months before I reach the full dose. But so far, no side effects, which is the purpose of doing it so slowly. Systrom seemed to connect his findings with autonomic dysfunction, and peripheral neuropathy seems to be associated with (if not a stronger connection) autonomic dysfunction. Maybe mestinon can be generally helpful for people with peripheral neuropathy. Fingers crossed.
“It was rather creepy, actually: during one week, the numbness and tingling starting in my foot, day by day progressed up to my knee.”
I had something similar. In less then 2 weeks numbness went up from my feet from almost none to reaching the spine between my shoulder blades and adding a strong blocked feeling to it.
With it went an increase in daily occurrences of short lived (1 to 2 second) of loss of control of the (all) leg muscles (temporary paralyzing where commands to move yielded absolutely nothing, not even an attempt to move). I had those before for about half a year but not at that frequency. That and the acceleration of it sure was scary!
I got a fairly quick reversal of it by starting circulation exercises. Mainly the neck circulation exercises proved to be very useful. Consult a good physical therapist when considering trying those however as doing these wrong can cause damage.
Peripheral Neuropathy is also found in GSD
I am so happy to read this article.
Three years ago I had all the symptoms of CFS after a viral infection. And There was one rhabdomyolysis.
All medical tests are normal.
Unfortunately, in Korea there is no Doctor who understands CFS properly.
I was diagnosed with mental disorders.
So I studied my own disease.
And ruled out similar diseases.
Finally I have found the presence of diseases in which both CFS symptoms and rhabdomyolysis can be explained.
Mitochondrial disease, glycogen storage disease, and lipid metabolism disorders.
I explained my hypotheses to doctors, but they ignored them.
Eventually I ordered a muscle biopsy and the result was mild myopathy.
However, there was no evidence of mitochondrial disorder, glycogen storage disease, or lipid metabolism disorders.
I received NGS PANEL test related to Exercise intolerance and many kind of myopathy.
The result was negative. No mutations were detected that were consistent with the symptoms.
I want to do WES Test, but the doctor will not do it.
Anyway this article gives me a lot of inspiration!
Good for you to be so pro-active Yun – and good luck in your search! If you find anything please let us know.
I did the Rhabdo test it was Negative so was the connective tissue panel & all porphyria was Negative
I was told today that the more common diagnosis in CFS looking for Glycogen Storage Disease is McArdle’s I think is types 5 not type 13 in Chronic Fatigue Syndrome…Those
patients with GSD 13 are just likely carriers of one Gene not two of them so time will tell which one & a biopsy is never diagnostic it is the Gene testing…I was also told
today to try to get the 18 gene panel done for GSD & also ask for a CK Creatinine blood test
Something just hit me on the site when she mentioned about Iron in the cells, not in the blood it could also explain Hypoglycemia events Systemic Nickel Allergy Syndrome. Doctors think it is only Contact but it is actually Systemic it could even
explain house aerosols reaction from the metal wet in can so maybe a subset do not have GSD13 or GSD5 maybe they have metal allergies to nickel, cobalt, stainless steel it is even in knives forks spoons pots pans roasting items even stainless steel
sinks door handles car home keys
Carbohydrate Metabolism Disorders & 32 gene panel for GSD cost £900.00 on the NHS here
Since we are talking about genes. This was an answer I replied on stenosis thread when asked about what I thought about calcium channel connections. Here is another thing to look into. This is not a simple, one possibility problem……it is complex with many possible connections.
Post copied and pasted:
https://www.healthrising.org/blog/2017/02/28/biomarker-aussies-chronic-fatigue-syndrome/
Note my comments from this blog and reference to another thread. I do think there is calcium channel issues.
https://www.sciencealert.com/one-of-the-biggest-myths-about-chronic-fatigue-syndrome-just-got-destroyed
And, for more technical info on this……
https://onlinelibrary.wiley.com/doi/full/10.1111/cei.12882
FYI — I have mutations on just about every snp you can test for TRMP3.
I still find GastroCrom to be one of my best helps – it not only moderates mast cells it is a mild calcium channel blocker. Since it is so expensive, I try not to use it all the time and when I do within a short time, it helps. Also 1/4 of my RX prescribed amount of Tramadol helps. It too has mild calcium channel blocking properties. (I tried regular calcium channel blockers, and that was too much. Maybe some other properties in these two things in combination is what helps.)
https://en.m.wikipedia.org/wiki/Calcium_channel_blocker
With more narrow and potentially hardend veins, vasodilation has been helpful to me. Also if there is too thick blood as with APS or Factor 8 and Collagen Binding which are also a problem with me….not only thinning blood but increasing diameter through which it flows, seems to be helpful. But…..since I have EDS, I must be careful how much I do this or POTS becomes more of an issue. I find external compression better than internal narrowing. Binders seem helpful in certain circumstances.
Issie
GSD 13 Glycogen Storage Disease low levels in enolase activity know to Cause Anemia in cell lines not found in blood test but enolase level can be tested & one was found to be 5% of normal levels in enolase ‘shortness of breath as well’
As we’ve discussed, I’m absolutely convinced that the CFS/ME diagnosis is catching a huge number of people with diagnosable inborn errors of metabolism. I actually wrote a whole blog post for you which is on hold until I can find an up-to-date geneticist to give advice on how to proceed with an adult onset metabolic investigation. The universe of IEM’s is vast and the standard exome tests, acylcarnitines, organic acids, lactate/pyruvate/ammonia etc. barely scratch the surface of what’s currently diagnostically possible. For exactly the reason discussed in this article I believe that adult onset IEM’s are vastly underdiagnosed. You won’t find what you aren’t looking for. Looking at the only large study of adult onset metabolic patients nearly all of them were eventually diagnosed after presenting with a sign specific enough to trigger testing. What’s important to note there is that many adult onset IEM’s will simply slowly progress into “normal” idiopathic disease processes such as heart disease and diabetes. The work that Dr Birch is doing is so important because we desperately need to separate IEM/CFS patients from non IEM/CFS patients to get good data out of the typical small size of ME/CFS research studies. It will be exciting to see what happens as bioinformatics allows for a deeper mining of an area that was considered a bust for ME/CFS not so long ago.
Hi Brian
Looking forward to that blog post when you are ready. Fereshtah, a Stanford researcher, told me that she is also picking up glucose storage issues at times in ME/CFS. She was happy to see Dr. Birch finding the same thing. It’s going to be very interesting…
Has she said anymore on this Glucose storage issue at Stanford & has the Genetic Team that went to Alabama to work more on Glycogen
Storage Disease out yet with any more findings? They mentioned the end of summer 2019…My other friend she & her son only ones sick
in the Family both have ‘aldolase B (HFI) Hereditary Fructose Intolerance’ & they also are Positive to ‘Baker Winegard Disease Fructose 1, 6’ they
also have Stiff Person Syndrome blood markers. They are now looking from Brain tumors like my other friend in the USA. Some on the UK
Group on HFI say do not eat any vegs any fruits ever with HFI they say eating certain fruits vegs is False & even in vitamins minerals medicines they eat High Meat clean diets
Cort – as I’m posting this reply 5 years after this article went up you may not see it, but I just want to let you know that I’m grateful for your work. I remember reading this, so it came immediately to mind this week when I got the disease screening results from a whole genome test that flagged me as most likely a carrier for GSDIII (high certainty) and GSDVI (medium certainty). If I am manifesting symptoms (it can happen in carriers of some diseases, so this is the hypothesis I’m going with) this is no doubt why I have done so well on a ketogenic regimen. (I wrote an article for you on my experiences.) As we have to figure this out for ourselves, this is leading me into more data collection to see if I can prove a connection through non-diabetic continuous glucose monitoring. It is really hard having to deal with medical professionals who see one particular symptom through their normal treatment lens when it needs to be addressed at a deeper level. I’m hanging in there!
Does anyone know where the photo above this text is from? “Problems with glycogen storage is another way to affect energy. Ron Tompkins will be assessing glycogen storage in ME/CFS muscle biopsies.”
I’m curious about the connection between thyroid hormone 1 and the TCA cycle, but cannot find this image anywhere.
Thank you for posting. It is worth pointing out that Dr. St. Amand ‘s protocol using Guaifenesin for Fibromyalgia has also been trialed in some patients with ME/CFS. Guaifenesin is the glyceryl ether of the botanical compound Guaicol. Guaicol has been used as a treatment for Polyglucosan disease, a rare condition that is characterized by…aberrant Glycogen storage.